A rare mitochondrial disease characterized by signs and symptoms within a phenotypic and metabolic spectrum that includes global developmental delay, hypotonia, intellectual disability, optic atrophy, axonal neuropathy, hypertrophic cardiomyopathy, lactic acidosis, and increased excretion of Krebs cycle intermediates. Other variable features are spasticity, seizures, ataxia, congenital cataract, and dysmorphic facial features. Age of onset is in the neonatal period or infancy.
A number sign (#) is used with this entry because of evidence that Harel-Yoon syndrome (HAYOS) is caused by heterozygous mutation in the ATAD3A gene (612316) on chromosome 1p36. One family with autosomal recessive inheritance has been reported. Description Harel-Yoon syndrome is a syndromic neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, truncal hypotonia, spasticity, and peripheral neuropathy. ... Clinical Features Harel et al. (2016) reported 5 unrelated children ranging from 23 months to 9 years of age with a syndromic neurodevelopmental disorder. All had severely delayed psychomotor development with intellectual disability and poor or absent speech.
A rare ciliopathy characterized by congenital moderate-to-severe deafness, retinitis pigmentosa developing in the first or second decade, and normal vestibular function. Congenital bilateral sensorineural hearing loss is mild to moderate in the low frequencies and severe to profound in the higher frequencies. Additional manifestations include night blindness, constricted visual field (tunnel vision), and later on decreased visual acuity sometimes ending with bare light perception.
A rare genetic neurological disorder characterized by motor developmental delay (in infancy), growth impairment and muscle weakness associated with myopathic abnormalities on muscle biopsy and EMG, as well as tremor, dysmetry, adiadochokinesia and walking disturbances associated with global or partial cerebral atrophy on brain MRI (particularly cerebellar vermis and hemispheres), with or without mild intellectual disability. Some patients also show pigmentary retinopathy.
A number sign (#) is used with this entry because of evidence that mitochondrial myopathy and ataxia (MMYAT) is caused by compound heterozygous mutation in the MSTO1 gene (617619) on chromosome 1q22. One family with a heterozygous mutation has been reported. Clinical Features Nasca et al. (2017) reported 2 sisters and an unrelated boy with mitochondrial myopathy and ataxia. The patients presented in the first year of life with delayed motor development and poor growth. The sisters were never able to walk independently, and had tremor, dysdiadochokinesia, and dysmetria associated with cerebellar hypoplasia on brain imaging. The younger sister had muscle weakness, pes cavus, and hyporeflexia. EMG showed a myopathic pattern, and muscle biopsies showed variability in fiber diameter with some dystrophic features.
Acheiria / ə ˈ k ɪər i ə / is the congenital absence of one or both hands. [1] [2] Causes [ edit ] It can occur in a number of situations which include: [1] Amniotic band syndrome , particularly if unilateral Cornelia de Lange syndrome Fetal hydantoin syndrome Incontinentia pigmenti References [ edit ] ^ a b Weerakkody, Dr Yuranga; et al.
A rare non-syndromic limb reduction defect characterized by the congenital total absence of the hand and wrist with no bony elements distal to the radius or ulna.
Hyperlipoproteinemia type 1 is an inherited condition that disrupts the normal breakdown of fats in the body, causing a large amount of fat to build up in the blood. This condition is characterized by inflammation of the pancreas ( pancreatitis ), abdominal pain, enlargement of the liver and spleen (hepatosplenomegaly), and small yellow skin lesions called eruptive xanthomas . Hyperlipoproteinemia type 1 is caused by mutations in the LPL gene. This condition is inherited in an autosomal recessive pattern. Treatment aims to control the symptoms through a low-fat diet.
A multigene panel that includes LPL and other genes of interest for the chylomicronemia syndrome (see Differential Diagnosis) may also be considered. ... Nomenclature Familial LPL deficiency is the most common form of the familial chylomicronemia syndrome, which was formerly known as "type 1 hyperlipoproteinemia." ... Differential Diagnosis Familial lipoprotein lipase (LPL) deficiency should be considered in young persons with the chylomicronemia syndrome, defined as abdominal pain, eruptive xanthomata, plasma triglyceride concentrations greater than 2000 mg/dL, and fasting lipemic plasma. ... Other than LPL deficiency, the chylomicronemia syndrome may be caused by biallelic pathogenic variants in apolipoprotein C-II ( APOC2 ), apolipoprotein A-V ( APOA5 ), lipase maturation factor 1 ( LMF1 ) or GPIHBP1 (see Table 2). ... Pancreatitis associated with the chylomicronemia syndrome is treated in the manner typical for other forms of pancreatitis.
Familial lipoprotein lipase deficiency is a rare genetic disorder is which a person lacks the enzyme lipoprotein lipase, a protein needed to break down fat molecules. Deficiency of this enzyme prevents affected individuals from properly digesting certain fats. This results in the accumulation of fatty droplets called chylomicrons in the blood and an increase in the blood concentration of triglycerides . Symptoms include episodes of abdominal pain, recurrent inflammation of the pancreas ( pancreatitis ), abnormal enlargement of the liver and/or spleen (hepatosplenomegaly), and the development of skin lesions known as erruptive xanthomas . Familial lipoprotein lipase deficiency is caused by changes (mutations) in the LPL gene.
It is managed by restricting fat in diet to less than 20 g/day. [2] The condition has also been called familial chylomicronemia syndrome, [3] chylomicronemia,. [4] : 533 chylomicronemia syndrome, [5] and hyperlipoproteinemia type Ia. [6] Contents 1 Signs and symptoms 1.1 Complications 2 Diagnosis 3 Treatment 3.1 Strict low fat diet and avoidance of simple carbohydrates 3.2 Lipid lowering drugs 3.3 Gene therapy 4 Incidence 5 See also 6 References 7 Further reading 8 External links Signs and symptoms [ edit ] The disease often presents in infancy with colicky pain, failure to thrive, and other symptoms and signs of the chylomicronemia syndrome. ... Typically, the plasma in a fasting blood sample appears creamy (plasma lactescence). [ medical citation needed ] Familial LPL deficiency should be considered in anyone with severe hypertriglyceridemia and the chylomicronemia syndrome. The absence of secondary causes of severe hypertriglyceridemia (like e.g. diabetes, alcohol, estrogen -, glucocorticoid -, antidepressant - or isotretinoin -therapy, certain antihypertensive agents , and paraproteinemic disorders) increases the possibility of LPL deficiency. ... External links [ edit ] Classification D ICD - 10 : E78 OMIM : 238600 MeSH : D008072 DiseasesDB : 4697 External resources MedlinePlus : 000408 GeneReviews : Familial Lipoprotein Lipase Deficiency Orphanet : 444490 v t e Inborn error of lipid metabolism : dyslipidemia Hyperlipidemia Hypercholesterolemia / Hypertriglyceridemia Lipoprotein lipase deficiency/Type Ia Familial apoprotein CII deficiency/Type Ib Familial hypercholesterolemia/Type IIa Combined hyperlipidemia/Type IIb Familial dysbetalipoproteinemia/Type III Familial hypertriglyceridemia/Type IV Xanthoma/Xanthomatosis Hypolipoproteinemia Hypoalphalipoproteinemia/HDL Lecithin cholesterol acyltransferase deficiency Tangier disease Hypobetalipoproteinemia/LDL Abetalipoproteinemia Apolipoprotein B deficiency Chylomicron retention disease Lipodystrophy Barraquer–Simons syndrome Other Lipomatosis Adiposis dolorosa Lipoid proteinosis APOA1 familial renal amyloidosis
Clinical Features Holt et al. (1939) first reported the familial occurrence of this syndrome. Boggs et al. (1957) described 3 affected sibs from a first-cousin mating. ... Gaudet et al. (2014) administered an inhibitor of APOC3 mRNA, called ISIS 304801, to treat 3 patients with familial chylomicronemia syndrome due to LPL deficiency and triglyceride levels ranging from 1,406 to 2,083 mg/dl (15.9-23.5 mM/l).
Familial lipoprotein lipase deficiency is an inherited condition that disrupts the normal breakdown of fats in the body, resulting in an increase of certain kinds of fats. People with familial lipoprotein lipase deficiency typically develop signs and symptoms before age 10, with one-quarter showing symptoms by age 1. The first symptom of this condition is usually abdominal pain, which can vary from mild to severe. The abdominal pain is often due to inflammation of the pancreas (pancreatitis). These episodes of pancreatitis begin as sudden (acute) attacks. If left untreated, pancreatitis can develop into a chronic condition that can damage the pancreas and, in rare cases, be life-threatening.
In a 21-month-old Japanese girl with physical and mental retardation, Omura et al. (1976) found excessive lysine in the urine, low lysine in the serum, and impaired intestinal absorption of lysine. They postulated a specific defect in lysine transport in the intestine and renal tubule. No information on the family was recorded. Growth - Growth retardation Neuro - Mental retardation Lab - Hyperlysinuria - Low serum lysine - Impaired lysine intestinal absorption - Intestinal lysine transport defect - Renal tubular lysine transport defect Inheritance - Autosomal recessive ▲ Close
Description IDDCDF is an autosomal recessive syndromic neurodevelopmental disorder characterized by globally impaired development with intellectual disability and speech delay, congenital cardiac malformations, and dysmorphic facial features. ... Clinical Features Stephen et al. (2018) reported 10 patients from 6 unrelated families with a similar syndromic neurodevelopmental disorder. The families were of different ethnic origins, but most were consanguineous and of Middle Eastern descent.
Congenital chylothorax is the most common cause of pleural effusion in neonates; it can occur primarily due to developmental anomalies of the lymphatic duct or can be associated with chromosomal anomalies (e.g. Noonan syndrome, Turner syndrome and Down syndrome), hydrops fetalis, mediastinal neuroblastoma and other congenital malformations.
Clinical Features Fox et al. (1998) described congenital chylothorax in 2 sibs, a female and a male. In the case of the female infant, the bilateral hydrothorax, more on the left with mediastinal shift, was discovered at 24 weeks' gestation. Bilateral thoracocentesis led to reexpansion of both lungs. Reaccumulation of fluid required bilateral pleuroamniotic shunts. The infant was delivered prematurely at 27 weeks' gestation and died at 12 hours of age. No evidence of congenital pulmonary lymphangiectasis was found at autopsy.
A rare genetic neurological disorder characterized by neonatal onset of rigidity and intractable seizures, with episodic jerking already beginning in utero . Affected infants have small heads, remain visually inattentive, do not feed independently, and make no developmental progress. Frequent spontaneous apnea and bradycardia usually culminate in cardiopulmonary arrest and death in infancy, although some cases were described with a milder clinical course and survival into childhood.
Biallelic mutations in the BRAT1 gene can also cause lethal neonatal rigidity and multifocal seizure syndrome (RMFSL; 614498), a more severe disorder with overlapping features.
A number sign (#) is used with this entry because of evidence that lethal neonatal rigidity and multifocal seizure syndrome (RMFSL) is caused by homozygous or compound heterozygous mutation in the BRAT1 gene (614506) on chromosome 7p22. ... Description Lethal neonatal rigidity and multifocal seizure syndrome is a severe autosomal recessive epileptic encephalopathy characterized by onset of rigidity and intractable seizures at or soon after birth. ... Inheritance The transmission pattern of a lethal neonatal rigidity and multifocal seizure syndrome in the Amish families reported by Puffenberger et al. (2012) was consistent with autosomal recessive inheritance. Molecular Genetics By homozygosity mapping followed by exome sequencing of 2 Amish patients from Pennsylvania with lethal neonatal rigidity and multifocal seizure syndrome, Puffenberger et al. (2012) identified a homozygous truncating mutation in the BRAT1 gene (614506.0001).
Multiple epiphyseal dysplasia (MED) is a group of disorders of cartilage and bone development, primarily affecting the ends of the long bones in the arms and legs (epiphyses). There are two types of MED, which are distinguished by their patterns of inheritance - autosomal dominant and autosomal recessive. Signs and symptoms may include joint pain in the hips and knees; early-onset arthritis; a waddling walk; and mild short stature as adults. Recessive MED may also cause malformations of the hands, feet, and knees; scoliosis; or other abnormalities. Most people are diagnosed during childhood, but mild cases may not be diagnosed until adulthood.
Some forms are mainly limited to the femoral epiphyses, while several other syndromes are characterized by the association of multiple epiphyseal dysplasia with other clinical manifestations such as myopia, deafness and facial dysmorphism.
A small vessel vasculitis characterized by neutrophilic inflammation predominantly limited to the superficial cutaneous postcapillary venules and without systemic vasculitis or glomerulonephritis. Typical presentation is of unifocal or multifocal palpable purpura on the lower extremities. Epidemiology The annual incidence of leukocytoclastic vasculitis in the United States is estimated at 1/220,000, of which 45% consist of cutaneous small vessel vasculitis. Clinical description The disease onset can occur at any age, and typically presents with non-blanching, palpable purpura and/or petechiae of the lower extremities with unifocal or multifocal distribution. The lesions may coalesce, ulcerate or be surrounded by hemorrhagic bullae.
Hypersensitivity vasculitis is an extreme reaction to a drug, infection, or foreign substance that leads to inflammation and damage to blood vessels of the skin. Signs and symptoms may include purple-colored spots and patches on the skin; skin lesions on the legs, buttocks, or trunk; blisters on the skin; hives ( urticaria ); and/or open sores with dead tissue (necrotic ulcers ). This condition is caused by an allergic reaction to a drug or other foreign substance. This condition usually goes away over time; but on occasion, people can have repeated episodes.
ARID1B -related disorder ( ARID1B -RD) constitutes a clinical continuum, from classic Coffin-Siris syndrome to intellectual disability with or without nonspecific dysmorphic features. ... See Coffin-Siris Syndrome for more information. Note: The information presented in the Coffin-Siris syndrome GeneReviews chapter includes information on individuals with CSS from a variety of genetic causes including ARID1B but is not specific to individuals with a pathogenic variant in ARID1B . ... AD = autosomal dominant; AR = autosomal recessive; CSS = Coffin-Siris syndrome; ID = intellectual disability; MOI = mode of inheritance; XL = X-linked 1. ... This latter characteristic is most likely to distinguish individuals with BOD syndrome from those with CSS, as the cognitive disability in CSS is nearly always moderate to severe. Inheritance appears to be autosomal dominant. Fetal alcohol syndrome (FAS). Small nails, prenatal and postnatal growth retardation, dysmorphic facial features, and cognitive disabilities may be seen in FAS.
Cleft palate can disrupt sucking-swallowing in newborns to varying degrees. In the non-syndromic forms, normal feeding is possible. In the syndromic forms, there is a risk of food aspiration. ... Etiology This embryopathy appears in the 7th to 12th week of pregnancy following an error in fusion of the palatine process. Non-syndromic clefts are believed to be caused by a combination of genetic and environmental factors. ... Differential diagnosis The presence of associated malformations allows for differentiation between isolated and syndromic forms. Differential diagnoses include hereditary syndromic forms (in 20% of cases), such as Pierre-Robin, Stickler, van der Woude and velocardiofacial syndromes (see these terms). ... Cleft palate can have functional consequences (morphological, phonetic, orthodontic, masticatory and auditory) that require management in a specialized health center. The syndromic forms, in particular a 22q11 deletion, have a poorer prognosis with more serious speech problems requiring complementary surgical treatment.
Christensen et al. (1992) found that in the Danish population, surgical files provided more than 95% ascertainment for cleft lip (with or without cleft palate) without associated malformations/syndromes. However, surgical files were a poor source for studying isolated cleft palate and could not be used to study the prevalence of associated malformations or syndromes.
Ovarian tumors occur with the Peutz-Jeghers syndrome (175200) and with the basal cell nevus syndrome (109400). ... No stigmata of basal cell nevus syndrome was found in the family reported by Dumont-Herskowitz et al. (1978).
A rare connective tissue disorder for which three subtypes exist, either related to the gene B4GALT7 , B3GALT6 or SLC39A13 , and for which the clinically overlapping characteristics include short stature (progressive in childhood), small joint hypermobility, skin hyperextensibility with soft, doughy skin especially on the hands and feet muscular hypotonia (ranging from congenitally severe to mild with later_onset), skeletal anomalies and, more variably, osteopenia, delayed motor development and bowing of the limbs. Gene-specific features, with variable presentation, are additionally observed in each subtype.
Cleft palate - stapes fixation - oligodontia is characterized by cleft soft palate, severe oligodontia of the deciduous teeth, absence of the permanent dentition, bilateral conductive deafness due to fixation of the footplate of the stapes, short halluces with a wide space between the first and second toes, and fusion of carpal and tarsal bones. It has been described in two sisters of Swedish extraction. An autosomal recessive mode of inheritance is likely. There have been no further descriptions in the literature since 1971.
In a sibship of Swedish extraction, Gorlin et al. (1971) observed 2 sisters with cleft soft palate, severe oligodontia of the deciduous teeth, no permanent dentition, bilateral conductive deafness due to fixation of the footplate of the stapes, short halluces with wide space between the first and second toes, and coalition of bones in the foot. Gorlin (1989) knew of no further cases. HEENT - Cleft soft palate - Severe oligodontia of deciduous teeth - No permanent dentition - Bilateral conductive deafness Inheritance - Autosomal recessive Skel - Short halluces - Wide space between first and second toes - Foot bone fusions ▲ Close
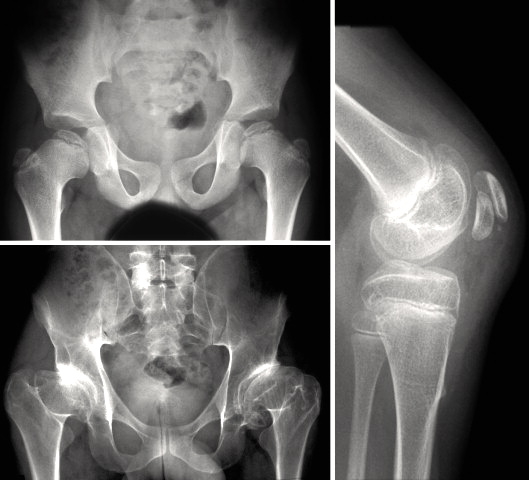
A rare, primary bone dysplasia characterized by proportional short stature, early cessation of bone growth, accelerated skeletal maturation, variable presence of early-onset osteoarthritis and osteochondritis dissecans, and normal endocrine evaluation. The variable dysmorphic features include mild to relative macrocephaly, frontal bossing, midfacial hypoplasia, flat nasal bridge, brachydactyly, broad thumbs, and lordosis.
Familial osteochondritis dissecans is a rare genetic skeletal disorder characterized clinically by abnormal chondro-skeletal development, disproportionate short stature and skeletal deformation mainly affecting the knees, hips, ankles and elbows with onset generally in late childhood or adolescence.
Escobar and Weaver (1978) suggested that these brothers had Aarskog syndrome (305400), and Berry et al. (1980) stated that they 'undoubtedly had Aarskog's syndrome.'
Familial osteochondritis dissecans is a condition that affects the joints and is associated with abnormal cartilage. Cartilage is a tough but flexible tissue that covers the ends of the bones at joints and is also part of the developing skeleton. A characteristic feature of familial osteochondritis dissecans is areas of bone damage (lesions) caused by detachment of cartilage and a piece of the underlying bone from the end of the bone at a joint. People with this condition develop multiple lesions that affect several joints, primarily the knees, elbows, hips, and ankles. The lesions cause stiffness, pain, and swelling in the joint. Often, the affected joint feels like it catches or locks during movement.
Osteochondritis dissecans is a joint condition that occurs when a piece of cartilage and the thin layer of bone beneath it, separates from the end of the bone. If the piece of cartilage and bone remain close to where they detached, they may not cause any symptoms. However, affected people may experience pain, weakness and/or decreased range of motion in the affected joint if the cartilage and bone travel into the joint space. Although osteochondritis dissecans can affect people of all ages, it is most commonly diagnosed in people between the ages of 10 and 20 years. In most cases, the exact underlying cause is unknown. Rarely, the condition can affect more than one family member (called familial osteochondritis dissecans); in these cases, osteochondritis dissecans is caused by changes (mutations) in the ACAN gene and is inherited in an autosomal dominant manner.