Angor animi Differential diagnosis acute coronary syndrome Angor animi (also referred to as angina animi , [1] [2] Gairdner's disease [2] and also angina pectoris sine dolore [2] ), in medicine , is a symptom defined as a patient's perception that they are in fact dying. Most cases of angor animi are found in patients suffering from acute coronary syndrome (cardiac related chest pain ) such as myocardial infarction . It is, however, occasionally found in patients suffering from other conditions. [1] Pheochromocytoma also can present with angor animi, accompanied by other symptoms that include; headache, profuse sweating, palpitations and characteristically a pounding severe headache. Irukandji syndrome is also another reported cause. [3] Angor animi is differentiated from a fear or desire for death, [1] since angor animi refers to a patient's actual and genuine belief that they are in fact dying. [1] Etymology [ edit ] The phrase is derived from the two Latin terms which it is composed of, namely angor and animi . ... "Carukia barnesi and the 'Irukndji Syndrome ' " (PDF) . www.marine-medic.com . ^ a b angor.
SYT1-associated neurodevelopmental disorder Other names Baker-Gordon Syndrome Specialty Medical genetics , Neurology SYT1 -associated neurodevelopmental disorder , also known as Baker-Gordon Syndrome , is a rare genetic disorder caused by mutations in the synaptotagmin-1 ( SYT1 ) gene. [1] Contents 1 Signs and symptoms 2 Genetics 3 Pathogenesis 4 Diagnosis 5 Management 6 History 7 References Signs and symptoms [ edit ] Patients present with neurodevelomental impairments and symptoms including: [ citation needed ] Infantile hypotonia Congenital ophthalmic abnormalities Childhood onset hyperkinetic movement disorder Sterotypical motor behaviour Moderate to profound developmental delay or intellectual disability Sleep disturbance Episodic agitation Epileptic seizures are not a feature of this disorder (despite abnormal EEG ) and head circumference is typically normal. ... Diagnosis is made through genetic testing with sequencing of the SYT1 gene. [ citation needed ] Management [ edit ] At present, only supportive management of symptoms is available as there is no known curative treatment for this condition. [ citation needed ] History [ edit ] The first case of SYT1 -associated neurodevelopmental disorder was described in 2015 [2] and it was classified as a syndrome in 2018. [1] [3] References [ edit ] ^ a b c Baker K, Gordon SL, Melland H, Bumbak F, Scott DJ, Jiang TJ, et al. ... PMID 25705886 . ^ "OMIM entry: Baker-Gordon Syndrome" .
IgA pemphigus Specialty Dermatology See also: List of target antigens in pemphigus IgA pemphigus is a subtype of pemphigus with two distinct forms: Subcorneal pustular dermatosis (also known as Sneddon–Wilkinson disease and pustulosis subcornealis) [1] is skin condition that is a rare, chronic, recurrent, pustular eruption characterized histopathologically by subcorneal pustules that contain abundant neutrophils. [2] [3] : 203 This is distinct from and not to be confused with subcorneal pustular dermatosis type of IgA pemphigus . Sneddon's syndrome , also known as Ehrmann-Sneddon syndrome, is also a different syndrome. [4] Intraepidermal neutrophilic IgA dermatosis is characterized histologically by intraepidermal bullae with neutrophils , some eosinophils , and acantholysis . [3] : 465 Contents 1 History 2 See also 3 References 4 External links History [ edit ] Early descriptions were made by Darrell Wilkinson , a British dermatologist. [5] See also [ edit ] Pemphigus List of cutaneous conditions List of conditions caused by problems with junctional proteins List of immunofluorescence findings for autoimmune bullous conditions References [ edit ] ^ RESERVED, INSERM US14 -- ALL RIGHTS. ... ISBN 0-7216-2921-0 . ^ Berlit, Peter. "Sneddon's Syndrome" . Orphanet . ^ "Munks Roll Details for Peter Edward Darrell Sheldon Wilkinson" . munksroll.rcplondon.ac.uk .
A rare autoimmune bullous skin disease characterized by painful and pruritic vesiculopustular eruptions resulting from circulating IgA antibodies against keratinocyte cell surface components. The lesions are typically found at the periphery of erythematous annular plaques and favor intertriginous regions. Histologically and immunologically, IgA pemphigus can be subdivided into subcorneal pustular dermatosis and intraepidermal neutrophilic IgA dermatosis.
Ischemic bone disease For other uses of "OCD", see OCD (disambiguation) . Osteochondritis dissecans A large flap lesion in the femur head typical of late stage Osteochondritis dissecans. In this case, the lesion was caused by avascular necrosis of the bone just under the cartilage. Pronunciation / ˌ ɒ s t i . oʊ k ɒ n ˈ d r aɪ t ɪ s ˈ d ɪ s ɪ k æ n z / Specialty Orthopedic surgery Osteochondritis dissecans ( OCD or OD ) is a joint disorder primarily of the subchondral bone in which cracks form in the articular cartilage and the underlying subchondral bone . [1] OCD usually causes pain during and after sports. In later stages of the disorder there will be swelling of the affected joint which catches and locks during movement.
Piezogenic papules Piezogenic papules on the heel of an individual with Ehlers–Danlos syndrome. Specialty Dermatology Painful fat herniation is foot pain caused by the herniation of fat through the thin fascial layers of the weight-bearing parts of the heel . [1] The herniation results in small bumps called piezogenic pedal papules [1] or piezogenic papules . [2] Though piezogenic papules are most commonly found on the heel, they can also be found on the wrist and palm. [3] Piezogenic papules are relatively common; in one population-based study, the prevalence was found to be 76%. [4] [3] They occur more frequently in runners, triathletes, and individuals exposed to long periods of standing. [4] They are also common in individuals with connective tissue disorders , especially Ehlers–Danlos syndrome . [5] [4] However, due to their preponderance in the general population, the presence of piezogenic papules alone does not automatically indicate the presence of Ehlers–Danlos syndrome. ... "The 2017 international classification of the Ehlers–Danlos syndromes" . Am J Med Genet C Semin Med Genet . 175 (1): 8–26. doi : 10.1002/ajmg.c.31552 .
Acrodysostosis syndrome Other names Arkless-Graham syndrome , [1] Maroteaux-Malamut syndrome [2] [3] Acrodysostosis is a rare congenital malformation syndrome which involves shortening of the interphalangeal joints of the hands and feet , intellectual disability in approximately 90% of affected children, and peculiar facies .
An acromelic dysplasia that is characterized by severe brachydactyly, peripheral dysostosis with facial dysostosis, nasal hypoplasia, and developmental delay. Epidemiology Less than 80 cases of Acrodysostosis (ACRDYS) have been reported in the literature to date. Clinical description Typical clinical features include severe peripheral dysostosis (short stature and brachydactyly affecting metacarpals, metatarsals and phalanges), facial dysostosis (broad face, widely spaced eyes and maxillonasal hypoplasia), and developmental delay. Advanced skeletal maturation, decreased vertebral interpedicular distance, and obesity are also frequently observed. Several features of acrodysostosis are similar to those present in patients with Albright's hereditary osteodystrophy (AHO) such as short stature, obesity and brachydactyly (in AHO, only 4th and 5th metacarpals and metatarsals).
At the age of 20, she suffered from recurrent carpal tunnel syndrome. The daughter showed cone-shaped epiphyses as in the mother. ... He also had dextrocardia, Kartagener syndrome (244400), and multiple orthopedic problems.
A number sign (#) is used with this entry because acrodysostosis-2 with or without hormone resistance (ACRDYS2) is caused by heterozygous mutation in the PDE4D gene (600129) on chromosome 5q12. Description Acrodysostosis-2 is a rare skeletal dysplasia characterized by brachydactyly, facial dysostosis, and spinal stenosis. Many patients have intellectual disability and some have hormone resistance (summary by Michot et al., 2012 and Lee et al., 2012). For a discussion of genetic heterogeneity of acrodysostosis, see ACRDYS1 (101800). Clinical Features Michot et al. (2012) reported 4 unrelated patients, ranging in age from 3 to 7 years, with acrodysostosis-2.
Acrodysostosis refers to a group of genetic disorders of bone growth. Common signs and symptoms include very short fingers and toes, underdeveloped facial bones, a small nose, and short stature. Many individuals with acrodysostosis have developmental delays and intellectual disability. Individuals with acrodysostosis additionally may have hormone resistance, which means that the body does not respond to the certain hormones. There are two types of this disorder, characterized by the presence or absence of hormone resistance and the underlying genetic cause.
Nitoiu et al. (2014) reviewed desmosome biology in cardiocutaneous syndromes and inherited skin disease, including discussion of the involvement of the DSP, PKP2, DSG2, DSC2, and JUP genes. ... The father subsequently had a 'flu-like' syndrome, heart failure, and biventricular dysfunction; 'active' myocarditis was found at endomyocardial biopsy.
Uhl anomaly is characterized by an almost complete absence of the myocardium in the right ventricle resulting in a thin walled nonfunctional right ventricle manifesting with cardiac arrhythmias and right ventricular failure. Cases of partial absence of right ventricular myocardium which remains asymptomatic or mildly symptomatic until adulthood have also been reported. Patients presenting with complete Uhl anomaly should be considered for cardiac transplantation.
Epidemiology Autosomal dominant cerebellar ataxia-deafness-narcolepsy syndrome (ADCA-DN) has been reported in more than 80 patients to date from Sweden, the United States, Italy, Brazil, Belgium, China, New Zealand, UK, Taiwan, Germany, and Canada.
Autosomal dominant cerebellar ataxia, deafness, and narcolepsy (ADCADN) is a nervous system disorder with signs and symptoms that usually begin in mid-adulthood and gradually get worse. People with ADCADN have difficulty coordinating movements (ataxia) and mild to moderate hearing loss caused by abnormalities of the inner ear (sensorineural deafness). Most have excessive daytime sleepiness (narcolepsy). Narcolepsy is typically accompanied by cataplexy, which is a sudden brief loss of muscle tone in response to strong emotion (such as excitement, surprise, or anger). These episodes of muscle weakness can cause an affected person to slump over or fall, which occasionally leads to injury. These characteristic signs and symptoms of ADCADN typically begin in a person's thirties.
A number sign (#) is used with this entry because of evidence that autosomal dominant cerebellar ataxia, deafness, and narcolepsy (ADCADN) is caused by heterozygous mutation in the DNMT1 gene (126375) on chromosome 19p13. Description ADCADN is an autosomal dominant neurologic disorder characterized by adult onset of progressive cerebellar ataxia, narcolepsy/cataplexy, sensorineural deafness, and dementia. More variable features include optic atrophy, sensory neuropathy, psychosis, and depression (summary by Winkelmann et al., 2012). Clinical Features Melberg et al. (1995) reported a 4-generation Swedish pedigree in which 5 individuals had cerebellar ataxia and sensorineural deafness. Four of the 5 patients also had narcolepsy. Among the 4, 2 had diabetes mellitus which was present in 2 otherwise healthy members of the family.
Some of these features can occur in distinct clusters on the phenotypic continuum: Boucher-Neuhäuser syndrome (cerebellar ataxia, chorioretinal dystrophy, and hypogonadotropic hypogonadism); Gordon Holmes syndrome (cerebellar ataxia, hypogonadotropic hypogonadism, and – to a variable degree – brisk reflexes); Oliver-McFarlane syndrome (trichomegaly, chorioretinal dystrophy, short stature, intellectual disability, and hypopituitarism); Laurence-Moon syndrome; and spastic paraplegia type 39 (SPG39) (upper motor neuron involvement, peripheral neuropathy, and sometimes reduced cognitive functioning and/or cerebellar ataxia). ... Cerebellar ataxia, hypogonadotropic hypogonadism, and (to a variable degree) brisk reflexes [Holmes 1907] Oliver-McFarlane syndrome. Trichomegaly, chorioretinal dystrophy, and congenital or childhood hypopituitarism [Hufnagel et al 2015, Kmoch et al 2015] Laurence-Moon syndrome. ... One family diagnosed with Laurence-Moon syndrome has been reported to have biallelic pathogenic variants in PNPLA6 [Hufnagel et al 2015]. ... In contrast, mutation of PNPLA6 is common in Boucher-Neuhäuser syndrome and Oliver-McFarlane syndrome: individuals in four of six families with Boucher-Neuhäuser syndrome and 11 of 12 families with Oliver-McFarlane syndrome had biallelic pathogenic variants in PNPLA6 [Synofzik et al 2014a, Hufnagel et al 2015, Kmoch et al 2015]. ... Marinesco-Sjögren syndrome is invariably associated with the combination of a cerebellar syndrome, chronic myopathy, and cataracts after age seven years [Krieger et al 2013]; myopathy and cataracts have not been reported in PNPLA6 -related disorders.
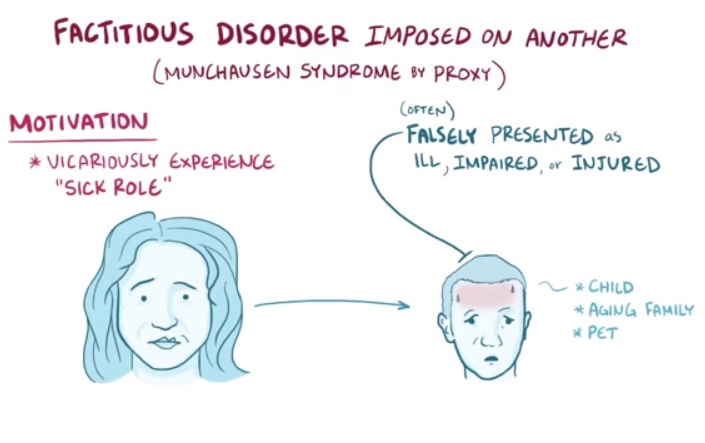
"Monsters in the Closet: Munchausen Syndrome by Proxy" (PDF) . CriticalCareNurse . ... Factitious Disorder/Munchhausen Syndrome . The 5-Minute Clinical Consult. 18th Edition. 2010. ... PMID 486971 . ^ a b Meadow R, Lennert T (October 1984). "Munchausen syndrome by proxy or Polle syndrome: which term is correct?" ... "Munchausen's syndrome". The Lancet . 1 (6650): 339–41. doi : 10.1016/S0140-6736(51)92313-6 . ... International Perspectives on Munchausen Syndrome by Proxy . Munchausen's syndrome by proxy: current issues in assessment, treatment and research .
Gonadal dysgenesis, including Turner syndrome , is the most common cause. Androgen insensitivity syndrome (Testicular feminization syndrome) Receptor abnormalities for hormones FSH and LH Specific forms of congenital adrenal hyperplasia Swyer syndrome Galactosaemia Aromatase deficiency Prader-Willi syndrome Male pseudo- hermaphroditism (about 1 in every 150,000 births) Müllerian agenesis /MRKH Syndrome Other intersexed conditions Hypothalamic: Kallmann syndrome Secondary Intrauterine adhesions ( Asherman's syndrome ) Pregnancy (most common cause) Anovulation Menopause Premature menopause Polycystic ovary syndrome (PCO-S) Drug-induced Breastfeeding Celiac disease Functional Hypothalamic : Exercise amenorrhoea, related to physical exercise , stress amenorrhoea, eating disorders and weight loss (obesity, anorexia nervosa , or bulimia ) Pituitary: Sheehan syndrome , hyperprolactinaemia , haemochromatosis Other central regulatory: hypothyroidism , hyperthyroidism , arrhenoblastoma Cause [ edit ] Low body weight [ edit ] Women who perform considerable amounts of exercise on a regular basis or lose a significant amount of weight are at risk of developing hypothalamic (or 'athletic') amenorrhoea. ... One example of this is MRKH (Mayer–Rokitansky–Küster–Hauser) syndrome, the second-most common cause of primary amenorrhoea. [32] The syndrome is characterized by Müllerian agenesis . ... The syndrome develops prenatally early in the development of the female reproductive system. ... Gonadal dysgenesis, often associated with Turner's Syndrome , or premature ovarian failure may also be to blame. ... Secondary amenorrhea may also be caused by outflow tract obstruction, often related to Asherman's Syndrome . Polycystic ovary syndrome can cause secondary amenorrhea, although the link between the two is not well understood.
Hormonal imbalance Many types of medical problems can cause hormonal imbalance, including: Polycystic ovary syndrome (PCOS). PCOS causes relatively high and sustained levels of hormones, rather than the fluctuating levels seen in the normal menstrual cycle. ... Examples include: Uterine scarring. Asherman's syndrome, a condition in which scar tissue builds up in the lining of the uterus, can sometimes occur after a dilation and curettage (D&C), cesarean section or treatment for uterine fibroids.
In severely raised intracranial pressure, the level of consciousness is decreased, the blood pressure rises, the heart rate falls and the patient assumes an abnormal posture. [2] Causes [ edit ] Cerebral venous sinus thrombosis is more common in particular situations. 85% of people have at least one of these risk factors: [2] Thrombophilia, a tendency to develop blood clots due to abnormalities in coagulation, e.g. factor V Leiden , deficiency of protein C , protein S or antithrombin , or related problems Nephrotic syndrome , a kidney problem causing protein loss in the urine Chronic inflammatory diseases, such as inflammatory bowel disease , lupus and Behçet's disease Pregnancy and puerperium (the period after giving birth) Particular blood disorders, especially polycythemia vera and paroxysmal nocturnal hemoglobinuria Use of estrogen-containing forms of hormonal contraception Meningitis and infections of the ear, nose and throat area such as mastoiditis and sinusitis Direct injury to the venous sinuses Medical procedures in the head and neck area Sickle cell anemia Dehydration , primarily in infants and children Homocystinuria Diagnosis [ edit ] The diagnosis may be suspected on the basis of the symptoms, for example the combination of headache, signs of raised intracranial pressure and focal neurological abnormalities, or when alternative causes of headache and neurological abnormalities, such as a subarachnoid hemorrhage , have been excluded. [2] Imaging [ edit ] CT venogram showing a filling defect in the sagittal sinus (black arrow) A dural venous sinus thrombosis of the transverse sinus. ... "Superior sagittal sinus thrombosis and pulmonary embolism: a syndrome rediscovered". Acta Neurol. Scand . 86 (4): 390–6. doi : 10.1111/j.1600-0404.1992.tb05106.x . ... PMID 7604412 . ^ Towbin A (1 May 1973). "The syndrome of latent cerebral venous thrombosis: its frequency and relation to age and congestive heart failure" . ... Retrieved 28 October 2007 . v t e Cerebrovascular diseases including stroke Ischaemic stroke Brain Anterior cerebral artery syndrome Middle cerebral artery syndrome Posterior cerebral artery syndrome Amaurosis fugax Moyamoya disease Dejerine–Roussy syndrome Watershed stroke Lacunar stroke Brain stem Brainstem stroke syndrome Medulla Medial medullary syndrome Lateral medullary syndrome Pons Medial pontine syndrome / Foville's Lateral pontine syndrome / Millard-Gubler Midbrain Weber's syndrome Benedikt syndrome Claude's syndrome Cerebellum Cerebellar stroke syndrome Extracranial arteries Carotid artery stenosis precerebral Anterior spinal artery syndrome Vertebrobasilar insufficiency Subclavian steal syndrome Classification Brain ischemia Cerebral infarction Classification Transient ischemic attack Total anterior circulation infarct Partial anterior circulation infarct Other CADASIL Binswanger's disease Transient global amnesia Haemorrhagic stroke Extra-axial Epidural Subdural Subarachnoid Cerebral/Intra-axial Intraventricular Brainstem Duret haemorrhages General Intracranial hemorrhage Aneurysm Intracranial aneurysm Charcot–Bouchard aneurysm Other Cerebral vasculitis Cerebral venous sinus thrombosis v t e Cardiovascular disease (vessels) Arteries , arterioles and capillaries Inflammation Arteritis Aortitis Buerger's disease Peripheral artery disease Arteriosclerosis Atherosclerosis Foam cell Fatty streak Atheroma Intermittent claudication Critical limb ischemia Monckeberg's arteriosclerosis Arteriolosclerosis Hyaline Hyperplastic Cholesterol LDL Oxycholesterol Trans fat Stenosis Carotid artery stenosis Renal artery stenosis Other Aortoiliac occlusive disease Degos disease Erythromelalgia Fibromuscular dysplasia Raynaud's phenomenon Aneurysm / dissection / pseudoaneurysm torso : Aortic aneurysm Abdominal aortic aneurysm Thoracic aortic aneurysm Aneurysm of sinus of Valsalva Aortic dissection Aortic rupture Coronary artery aneurysm head / neck Intracranial aneurysm Intracranial berry aneurysm Carotid artery dissection Vertebral artery dissection Familial aortic dissection Vascular malformation Arteriovenous fistula Arteriovenous malformation Telangiectasia Hereditary hemorrhagic telangiectasia Vascular nevus Cherry hemangioma Halo nevus Spider angioma Veins Inflammation Phlebitis Venous thrombosis / Thrombophlebitis primarily lower limb Deep vein thrombosis abdomen Hepatic veno-occlusive disease Budd–Chiari syndrome May–Thurner syndrome Portal vein thrombosis Renal vein thrombosis upper limb / torso Mondor's disease Paget–Schroetter disease head Cerebral venous sinus thrombosis Post-thrombotic syndrome Varicose veins Gastric varices Portacaval anastomosis Caput medusae Esophageal varices Hemorrhoid Varicocele Other Chronic venous insufficiency Chronic cerebrospinal venous insufficiency Superior vena cava syndrome Inferior vena cava syndrome Venous ulcer Arteries or veins Angiopathy Macroangiopathy Microangiopathy Embolism Pulmonary embolism Cholesterol embolism Paradoxical embolism Thrombosis Vasculitis Blood pressure Hypertension Hypertensive heart disease Hypertensive emergency Hypertensive nephropathy Essential hypertension Secondary hypertension Renovascular hypertension Benign hypertension Pulmonary hypertension Systolic hypertension White coat hypertension Hypotension Orthostatic hypotension
Najm type X-linked intellectual deficit is a rare cerebellar dysgenesis syndrome characterized by variable clinical manifestations ranging from mild intellectual deficit with or without congenital nystagmus, to severe cognitive impairment associated with cerebellar and pontine hypoplasia/atrophy and abnormalities of cortical development. Epidemiology Prevalence of this rare neurological syndrome is unknown. Up to 35 families have been reported to date. ... Etiology Point mutations and deletions in the CASK gene (Xp11.4) have been found in patients with this syndrome. Diagnostic methods Magnetic resonance imaging (MRI) generally shows pontocerebellar hypoplasia/atrophy and simplified cortical gyri.
A number sign (#) is used with this entry because of evidence that the MICPCH syndrome is caused by heterozygous mutation or deletion in the CASK gene (300172) on chromosome Xp11. Missense variants in the CASK gene have been shown to cause a milder mental retardation syndrome, sometimes including nystagmus, most often in males (FGS4; 300422). ... Saitsu et al. (2012) reported 2 unrelated Japanese males ascertained for severe early-onset epileptic encephalopathy consistent with a diagnosis of Ohtahara syndrome who were found to carry hemizygous loss-of-function mutations in the CASK gene.
Some researchers have suggested that a condition called FG syndrome 4 may also be caused by mutations in the CASK gene, but it seems that the only family described with this syndrome may have actually had XL-ID, with or without nystagmus.
Specialty Medical genetics Intellectual disability and microcephaly with pontine and cerebellar hypoplasia (MICPCH), also known as mental retardation, X-linked, syndromic, Najm type (MRXSNA), is a rare X-linked dominant genetic disorder of infants characterised by intellectual disability and pontocerebellar hypoplasia .
Cystic hygromas can occur as an isolated finding or in association with other birth defects as part of a syndrome (chromosomal abnormalities or syndromes caused by gene mutations). ... The majority of prenatally diagnosed cystic hygromas are associated with Turner syndrome or other chromosomal abnormalities like trisomy 21 .