Differential diagnosis Differential diagnoses include the other forms of OCA and X-linked recessive ocular albinism (XLOA) as well as syndromes with albinism as a feature such as Hermansky-Pudlak syndromes 1-7, Chediak-Higashi syndrome , Griscelli syndrome, and Waardenburg syndrome type II.
Disorders to Consider in the Differential Diagnosis of OCA4 View in own window Disorder Gene(s) MOI Clinical Features of the Differential Disorder Overlapping w/OCA4 Distinguishing from OCA4 OCA1 (OMIM 203100, 606952) TYR AR Oculocutaneous albinism May have poorer visual acuity OCA2 (OMIM 203200) OCA2 AR NA OCA3 (OMIM 203290) TYRP1 AR Reddish hair & freckled skin; only seen in individuals of African, Pakistani, German, Indian, & Japanese heritage OCA5 (OMIM 615312) Unknown (candidate region is on 4q24) AR Golden hair OCA6 (OMIM 113750) SLC24A5 AR Indistinguishable due to functional similarity of SLC45A2 (OCA4) & SLC24A5 OCA7 (OMIM 615179) LRMDA (formerly C10orf11 ) AR Relatively severe impaired visual acuity Hermansky-Pudlak syndrome (HPS) HPS1 AP3B1 ( HPS2 ) HPS3 HPS4 HPS5 HPS6 DTNBP1 ( HPS7 ) BLOC1S3 ( HPS8 ) BLOC1S6 AP3D1 (HPS10) AR Bleeding tendency, platelet dense granules; granulomatous colitis & interstitial pneumonia in HPS1 & HPS4; immunodeficiency & hemophagocytic syndrome in HPS2 & HPS10 OA1 GPR143 XL Ocular albinism Coloring may appear normal but some may have mild hypopigmentation of skin & hair compared to family members.
A number sign (#) is used with this entry because oculocutaneous albinism type IV (OCA4) is caused by homozygous or compound heterozygous mutation in the MATP gene (SLC45A2; 606202) on chromosome 5p13. For a general phenotypic description and a discussion of genetic heterogeneity of oculocutaneous albinism, see OCA1 (203100). Clinical Features Newton et al. (2001) reported a Turkish patient with generalized hypopigmentation and ocular abnormalities consistent with OCA. He had white hair, pale skin, and translucent blue-gray irides. The phenotype was reminiscent of the relatively mild OCA2 (203200). Inagaki et al. (2004) reported Japanese patients with OCA4. Hair color ranged from white to yellow to brown, and iris color ranged from blue to brown.
Metastatic calcification [ edit ] Metastatic calcification involves a systemic calcium excess imbalance, which can be caused by hypercalcemia , kidney failure , milk-alkali syndrome , lack or excess of other minerals, or other causes. ... See also [ edit ] Calcinosis cutis Dermatomyositis Fahr's syndrome Hyperphosphatemia Primrose syndrome Scleroderma References [ edit ] External links [ edit ] Classification D ICD - 9-CM : 275.4 MeSH : D002114 SNOMED CT : 6595006 v t e Electrolyte imbalances Sodium High Salt poisoning Low Hypotonic Isotonic Cerebral salt-wasting syndrome Potassium High Low Chloride High Low Calcium High Low Symptoms and signs Chvostek sign Trousseau sign Milk-alkali syndrome Disorders of calcium metabolism Calcinosis ( Calciphylaxis , Calcinosis cutis ) Calcification ( Metastatic calcification , Dystrophic calcification ) Familial hypocalciuric hypercalcemia Phosphate High Low Magnesium High Low This article about a disease , disorder, or medical condition is a stub .
Some patients may develop lymphatic and hepatic malignancies, usually as late complications. MC syndrome may be associated with numerous infectious or immunological diseases. ... Moreover, MC may be associated with other infectious agents or immunological disorders, such as human immunodeficiency virus (HIV) infection or primary Sjogren syndrome. Diagnostic methods Diagnosis is based on clinical and laboratory findings. ... Differential diagnosis Differential diagnoses include a wide range of systemic, infectious and neoplastic disorders, mainly autoimmune hepatitis, primary Sjögren syndrome, B-cell lymphomas, and rheumatoid arthritis. ... Use of immunomodifiers, immunosuppressor and plasmapheresis may play a major role in HCV-negative MC ('essential' MC syndrome) or in clinically active/relapsed MC that can be observed also after HCV eradication in some patients. ... Similarly, timely treatment of more severe manifestations of the MC syndrome can prevent progression of organ damage.
This article needs additional citations for verification . Please help improve this article by adding citations to reliable sources . Unsourced material may be challenged and removed. Find sources: "Cryoglobulinemic vasculitis" – news · newspapers · books · scholar · JSTOR ( April 2009 ) ( Learn how and when to remove this template message ) Cryoglobulinemic vasculitis Other names Essential mixed cryoglobulinemia, Primary cryoglobulinemia [1] Cryoglobulinemic vasculitis is inherited in an autosomal dominant manner Specialty Dermatology Cryoglobulinemic vasculitis is a form of inflammation affecting the blood vessels caused by the deposition of abnormal proteins called cryoglobulins in the blood vessels. Cryoglobulinemic vasculitis affects the skin and causes a rash in roughly 15% of people with detectable circulating cryoglobulin proteins. [2] : 835 Additionally, the kidneys may be affected by this form of vasculitis resulting in membranoproliferative glomerulonephritis . Fevers , painful muscles and joints , and peripheral nerve damage are other common manifestations of cryoglobulinemic vasculitis. Due to deposition of complement (in particular, C4), low levels of circulating complement factors may be seen.
A number sign (#) is used with this entry because cerebral creatine deficiency syndrome-3 (CCDS3), also known as arginine:glycine amidinotransferase (AGAT) deficiency, is caused by homozygous mutation in the GATM gene (602360) on chromosome 15q21. Description Cerebral creatine deficiency syndrome-3 is an autosomal recessive disorder characterized by developmental delay/regression, mental retardation, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain (Schulze, 2003). ... For a general phenotypic description and a discussion of genetic heterogeneity of cerebral creatine deficiency syndrome, see CCDS1 (300352). Clinical Features Bianchi et al. (2000) reported 2 female sibs, aged 4 and 6 years, with mental retardation and severe creatine deficiency in the brain. ... Edvardson et al. (2010) reported 2 sibs, born of unrelated Yemenite Jewish parents, with cerebral creatine deficiency syndrome. Both showed delayed psychomotor development and failure to thrive in infancy. ... In 2 sibs, born of unrelated Yemenite Jewish parents, with cerebral creatine deficiency syndrome-3, Edvardson et al. (2010) identified a homozygous truncating mutation in the GATM gene (602360.0002).
Contents 1 Signs and symptoms 2 Genetics 3 Diagnosis 4 Treatment 5 References 6 External links Signs and symptoms [ edit ] As with other cerebral creatine deficiency syndromes, individuals affected with AGAT deficiency are intellectually disabled and can have seizures . ... References [ edit ] ^ a b c d e "Creatine Deficiency Syndromes" . Creatine Deficiency Syndrome . ... Retrieved 2018-09-26 . ^ "CEREBRAL CREATINE DEFICIENCY SYNDROME 3; CCDS3" . Johns Hopkins University . Retrieved 2018-10-11 . ^ a b c Schulze, Andreas (2009). "Creatine Deficiency Syndromes". In Sarafoglou, Kiriakie; Hoffmann, Georg F.; Roth, Karl S.
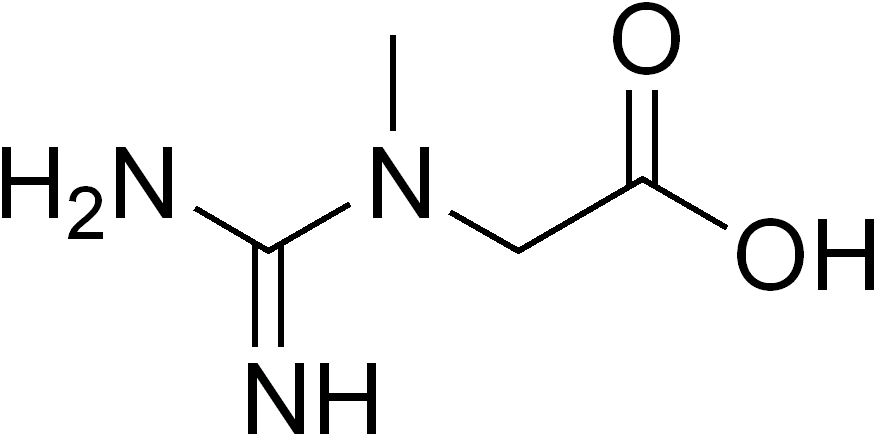
L-Arginine:glycine amidinotransferase (AGAT) deficiency is a very rare type of creatine deficiency sydrome characterized by global developmental delay, intellectual disability, and myopathy. Epidemiology Less than 20 patients have been described with AGAT deficiency to date. Clinical description AGAT deficiency is characterized by global developmental delay, appearing in infancy, which can be associated with language impairment and autistic behavior in some, as well as a mild to moderate intellectual disability. Progressive muscle weakness and fatigability have been reported in older patients. Seizures and failure to thrive have also been described. If creatine supplementation is administered early enough, psychomotor delay may be avoided.
Arginine:glycine amidinotransferase deficiency is an inherited disorder that primarily affects the brain. People with this disorder have mild to moderate intellectual disability and delayed speech development. Some affected individuals develop autistic behaviors that affect communication and social interaction. They may experience seizures, especially when they have a fever. Children with arginine:glycine amidinotransferase deficiency may not gain weight and grow at the expected rate (failure to thrive), and have delayed development of motor skills such as sitting and walking. Affected individuals may also have weak muscle tone and tend to tire easily.
L-arginine:glycine amidinotransferase (AGAT) deficiency is a rare condition that primarily affects the brain. People with AGAT deficiency generally have mild to moderate intellectual disability. Other signs and symptoms may include seizures, delayed language development, muscle weakness, failure to thrive , autistic behaviors , and delayed motor milestones (i.e. walking, sitting). AGAT deficiency is caused by changes (mutations) in the GATM gene and is inherited in an autosomal recessive manner. Treatment of AGAT deficiency is focused on increasing cerebral creatine levels and generally consists of supplementation with creatine monohydrate .
The word hwabyung is composed of hwa (the Sino-Korean word 火 for "fire" which can also contextually mean "anger") and byung (the Sino-Korean word 病 for "syndrome" or "illness"). [5] It may also be called ulhwabyeong ( 鬱火病 ), literally "depression anger illness". ... "Symptoms to Use for Diagnostic Criteria of Hwa-Byung, an Anger Syndrome" . Psychiatry Investig . 6 (1): 7–12. doi : 10.4306/pi.2009.6.1.7 . ... Identifying and treating the culture-bound syndrome of Hwa-Byung among older Korean immigrant women: Recommendations for practitioners. ... Symptoms to use for Diagnostic Criteria of Hwa-Byung, an Anger Syndrome. Psychiatry Investig. 2009 March; 6(1): 7–12. Published online 2009 March 31.doi: 10.4306/pi.2009.6.1.7 Hwa-byung: Culture-related Syndrome Hwabyung in Korea: Culture and Dynamic Analysis
Immersion foot Trench foot as seen on an unidentified soldier during World War I Specialty Dermatology Immersion foot syndromes are a class of foot injury caused by water absorption in the outer layer of skin. [1] [2] There are different subclass names for this condition based on the temperature of the water to which the foot is exposed. ... "The role of temperature in tropical immersion foot syndrome" . The Journal of the American Medical Association . 202 (6): 546–549. doi : 10.1001/jama.1967.03130190152032 . ... External links [ edit ] Classification D ICD - 10 : T69.0 ICD - 9-CM : 991.4 MeSH : D007102 DiseasesDB : 31219 v t e Consequences of external causes Temperature Elevated Hyperthermia Heat syncope Reduced Hypothermia Immersion foot syndromes Trench foot Tropical immersion foot Warm water immersion foot Chilblains Frostbite Aerosol burn Cold intolerance Acrocyanosis Erythrocyanosis crurum Radiation Radiation poisoning Radiation burn Chronic radiation keratosis Eosinophilic, polymorphic, and pruritic eruption associated with radiotherapy Radiation acne Radiation-induced cancer Radiation recall reaction Radiation-induced erythema multiforme Radiation-induced hypertrophic scar Radiation-induced keloid Radiation-induced morphea Air Hypoxia / Asphyxia Barotrauma Aerosinusitis Decompression sickness High altitude Altitude sickness Chronic mountain sickness Death zone HAPE HACE Food Starvation Maltreatment Physical abuse Sexual abuse Psychological abuse Travel Motion sickness Seasickness Airsickness Space adaptation syndrome Adverse effect Hypersensitivity Anaphylaxis Angioedema Allergy Arthus reaction Adverse drug reaction Other Electrical injury Drowning Lightning injuries Ungrouped skin conditions resulting from physical factors Dermatosis neglecta Pinch mark Pseudoverrucous papules and nodules Sclerosing lymphangitis Tropical anhidrotic asthenia UV-sensitive syndrome environmental skin conditions Electrical burn frictional/traumatic/sports Black heel and palm Equestrian perniosis Jogger's nipple Pulling boat hands Runner's rump Surfer's knots Tennis toe Vibration white finger Weathering nodule of ear Wrestler's ear Coral cut Painful fat herniation Uranium dermatosis iv use Skin pop scar Skin track Slap mark Pseudoacanthosis nigricans Narcotic dermopathy
Handigodu syndrome Specialty Orthopedic Handigodu syndrome is a rare and painful osteoarthritic disorder endemic to the Malnad region in the state of Karnataka , India . [1] Also known as Handigodu Joint Disease , it derives its name from the village of Handigodu in the Sagara taluk of the Shimoga district of Karnataka where it was first noticed. ... The study reported that Handigodu syndrome is a syndrome of familial spondyloepi(meta)physeal dysplasia . [5] It is inherited as an autosomal dominant trait. [5] All the presentations of the varied manifestation of the disease could be explained as being caused by defective development of bones as a result of monogenic disorder . [5] History [ edit ] Handigodu syndrome was first noticed by H M Chandrashekhar in the village of Handigodu during the years 1974-75. [4] Four patients who complained of severe joint and hip pain were admitted to the General Hospital at Sagar .
Spondyloepimetaphyseal dysplasia, Handigodu type is a rare, genetic, primary bone dysplasia disorder characterized by three distinct phenotypes, namely: 1) patients of average height with painful, osteoarthritic changes of the hip joints and no spinal abnormalities, 2) short-statured patients with predominantly truncal shortening, arm span exceeding height, dysplastic changes of hips and varying degrees of platyspondyly, and 3) patients with dwarfism, various associated skeletal abnormalities (particularly of the knees and hands) and severe epiphyseal dysplasia (of hips, knees, hands, wrists) associated with significant platyspondyly. Most patients cannot walk long distances, and many have decreased joint spaces, as well as sclerotic and cystic changes on imaging.
Description Handigodu disease is a autosomal dominant spondyloepimetaphyseal dysplasia prevalent in a few villages of 2 districts of the state of Karnataka in southern India (Agarwal et al., 1994). Clinical Features Handigodu disease is a progressive disorder of the skeletal system, predominantly involving the hip joints and spine, with onset in late childhood or young adulthood, which ultimately leads to significant handicap with flexion deformity of hips, lumbar lordosis, waddling gait, and difficulty in squatting and sitting cross-legged. Three main groups of clinical presentations have been identified. The first consists of average height individuals with predominantly osteoarthritic changes of the hip joints. Most affected persons belong to a second group, consisting of short-statured individuals in whom arm span exceeds height. The third group presents with dwarfism and in these persons involvement of the spine is more severe.
PMID 316945 . v t e Physiology of the visual system Vision Accommodation Gaze Intraocular pressure Visual field Color vision Color blindness Achromatopsia Köllner's rule Opponent process Dichromacy Monochromacy Pentachromacy Tetrachromacy Trichromacy v t e Diseases of the human eye Adnexa Eyelid Inflammation Stye Chalazion Blepharitis Entropion Ectropion Lagophthalmos Blepharochalasis Ptosis Blepharophimosis Xanthelasma Ankyloblepharon Eyelash Trichiasis Madarosis Lacrimal apparatus Dacryoadenitis Epiphora Dacryocystitis Xerophthalmia Orbit Exophthalmos Enophthalmos Orbital cellulitis Orbital lymphoma Periorbital cellulitis Conjunctiva Conjunctivitis allergic Pterygium Pseudopterygium Pinguecula Subconjunctival hemorrhage Globe Fibrous tunic Sclera Scleritis Episcleritis Cornea Keratitis herpetic acanthamoebic fungal Exposure Photokeratitis Corneal ulcer Thygeson's superficial punctate keratopathy Corneal dystrophy Fuchs' Meesmann Corneal ectasia Keratoconus Pellucid marginal degeneration Keratoglobus Terrien's marginal degeneration Post-LASIK ectasia Keratoconjunctivitis sicca Corneal opacity Corneal neovascularization Kayser–Fleischer ring Haab's striae Arcus senilis Band keratopathy Vascular tunic Iris Ciliary body Uveitis Intermediate uveitis Hyphema Rubeosis iridis Persistent pupillary membrane Iridodialysis Synechia Choroid Choroideremia Choroiditis Chorioretinitis Lens Cataract Congenital cataract Childhood cataract Aphakia Ectopia lentis Retina Retinitis Chorioretinitis Cytomegalovirus retinitis Retinal detachment Retinoschisis Ocular ischemic syndrome / Central retinal vein occlusion Central retinal artery occlusion Branch retinal artery occlusion Retinopathy diabetic hypertensive Purtscher's of prematurity Bietti's crystalline dystrophy Coats' disease Sickle cell Macular degeneration Retinitis pigmentosa Retinal haemorrhage Central serous retinopathy Macular edema Epiretinal membrane (Macular pucker) Vitelliform macular dystrophy Leber's congenital amaurosis Birdshot chorioretinopathy Other Glaucoma / Ocular hypertension / Primary juvenile glaucoma Floater Leber's hereditary optic neuropathy Red eye Globe rupture Keratomycosis Phthisis bulbi Persistent fetal vasculature / Persistent hyperplastic primary vitreous Persistent tunica vasculosa lentis Familial exudative vitreoretinopathy Pathways Optic nerve Optic disc Optic neuritis optic papillitis Papilledema Foster Kennedy syndrome Optic atrophy Optic disc drusen Optic neuropathy Ischemic anterior (AION) posterior (PION) Kjer's Leber's hereditary Toxic and nutritional Strabismus Extraocular muscles Binocular vision Accommodation Paralytic strabismus Ophthalmoparesis Chronic progressive external ophthalmoplegia Kearns–Sayre syndrome palsies Oculomotor (III) Fourth-nerve (IV) Sixth-nerve (VI) Other strabismus Esotropia / Exotropia Hypertropia Heterophoria Esophoria Exophoria Cyclotropia Brown's syndrome Duane syndrome Other binocular Conjugate gaze palsy Convergence insufficiency Internuclear ophthalmoplegia One and a half syndrome Refraction Refractive error Hyperopia Myopia Astigmatism Anisometropia / Aniseikonia Presbyopia Vision disorders Blindness Amblyopia Leber's congenital amaurosis Diplopia Scotoma Color blindness Achromatopsia Dichromacy Monochromacy Nyctalopia Oguchi disease Blindness / Vision loss / Visual impairment Anopsia Hemianopsia binasal bitemporal homonymous Quadrantanopia subjective Asthenopia Hemeralopia Photophobia Scintillating scotoma Pupil Anisocoria Argyll Robertson pupil Marcus Gunn pupil Adie syndrome Miosis Mydriasis Cycloplegia Parinaud's syndrome Other Nystagmus Childhood blindness Infections Trachoma Onchocerciasis v t e Color topics Red Orange Yellow Green Cyan Blue Indigo Violet Purple Magenta Pink Brown White Gray Black Color science Color physics Electromagnetic spectrum Light Rainbow Visible Spectral colors Chromophore Structural coloration Animal coloration Color of chemicals Water On Vision and Colours Metamerism Spectral power distribution Color perception Color vision Color blindness Achromatopsia test Tetrachromacy Color constancy Color term Color depth Color photography Spot color Color printing Web colors Color mapping Color code Color management Chrominance False color Chroma key Color balance Color cast Color temperature Eigengrau The dress Color psychology Color symbolism Color preferences Lüscher color test Kruithof curve Political color National colors Chromophobia Chromotherapy Color philosophy Color space Color model additive subtractive Color mixing Primary color Secondary color Tertiary color (intermediate) Quaternary color Quinary color Aggressive color (warm) Receding color (cool) Pastel colors Color gradient Color scheme Color tool Monochromatic colors Complementary colors Analogous colors Achromatic colors (Neutral) Polychromatic colors Impossible colors Light-on-dark Tinctures in heraldry Color theory Chromaticity diagram Color solid Color wheel Color triangle Color analysis (art) Color realism (art style) Color terms Basic terms Blue Green Red Yellow Pink Purple Orange Black Gray White Brown Cultural differences Linguistic relativity and the color naming debate Blue–green distinction in language Color history Color in Chinese culture Traditional colors of Japan Human skin color Color dimensions Hue Dichromatism Colorfulness (chroma and saturation) Tints and shades Lightness (tone and value) Grayscale Color organizations Pantone Color Marketing Group Color Association of the United States International Colour Authority International Commission on Illumination (CIE) International Color Consortium International Colour Association Lists List of colors: A–F List of colors: G–M List of colors: N–Z List of colors (compact) List of colors by shade List of color palettes List of color spaces List of Crayola crayon colors history Color chart List of RAL colors List of web colors Related Vision Digital image processing Multi-primary color display Quattron Qualia Lighting Local color (visual art) Category Index This article about an ophthalmic disease is a stub .
17 alpha(α)-hydroxylase/17,20-lyase deficiency is a condition that affects the function of certain hormone-producing glands called the gonads (ovaries in females and testes in males) and the adrenal glands. The gonads direct sexual development before birth and during puberty and are important for reproduction. The adrenal glands, which are located on top of the kidneys, regulate the production of certain hormones, including those that control salt levels in the body. People with 17α-hydroxylase/17,20-lyase deficiency have an imbalance of many of the hormones that are made in these glands. 17α-hydroxylase/17,20-lyase deficiency is one of a group of disorders, known as congenital adrenal hyperplasias, that impair hormone production and disrupt sexual development and maturation. Hormone imbalances lead to the characteristic signs and symptoms of 17α-hydroxylase/17,20-lyase deficiency, which include high blood pressure (hypertension), low levels of potassium in the blood (hypokalemia), and abnormal sexual development.
The authors concluded that basal progesterone measurement is a useful marker of P450c17 deficiency and that its use should reduce the misdiagnosis of this deficiency in patients presenting with male pseudohermaphroditism, primary or secondary amenorrhea, and mineralocorticoid excess syndrome. Combined Partial 17-alpha-Hydroxylase/17,20-Lyase Deficiency Oshiro et al. (1995) reported a 32-year-old Japanese woman with combined partial 17-alpha-hydroxylase/17,20-lyase deficiency caused by mutation in the CYP17A1 gene (609300.0008). ... GU - Ambiguous genitalia - Primary amenorrhea - Male pseudohermaphroditism Inheritance - Autosomal recessive Metabolic - Hypertension - Hypokalemic alkalosis Lab - 17-alpha-hydroxylase deficiency - ACTH increased - FSH increased Thorax - Gynecomastia Endo - Adrenogenital syndrome ▲ Close
46,XY disorder of sex development due to isolated 17,20-lyase deficiency is a rare disorder of sex development due to reduced 17,20-lyase activity that affects individuals with 46,XY karyotype and is characterized by ambiguous external genitalia, including micropenis, perineal hypospadias, bifid scrotum, cryptorchidism, and a blind vaginal pouch. Blood pressure and electrolytes are normal whilst hormonal investigations show normal basal and stimulated levels of cortisol, and low basal and stimulated androgen levels.
A very rare form of congenital adrenal hyperplasia (CAH) characterized by glucocorticoid deficiency, hypergonadotrophic hypogonadism and severe hypokalemic hypertension. Epidemiology It accounts for approximately 1% of all CAH cases. The prevalence is therefore around 1/1,000,000. Clinical description Both a sex steroid and glucocorticoid deficiency are present. Common manifestations include undervirilization in males, primary amenorrhea in females and lack of pubertal development in both sexes. Hypertension, often accompanied by hypokalemia, can also develop due to the mineralocorticoid excess seen in this disease.
With viral pneumonia, samples are taken from the upper and/or lower respiratory tracts. [3] The samples can then be run through polymerase chain reaction (PCR), allowing for amplification of the virus as that allows better detection if present in the sample. [4] Other ways for a diagnosis to be obtained is by ordering a chest x-ray , blood tests, pulse oximetry , and a medical/family history to see if there are any known risks or previous exposures to a person with viral pneumonia. [4] If the person is in serious condition and in the hospital there are more invasive studies that can be run to diagnosis the person. [4] Cause [ edit ] Common causes of viral pneumonia are: Influenza virus A and B [5] Respiratory syncytial virus (RSV) [5] Human parainfluenza viruses (in children) [5] Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [6] Rarer viruses that commonly result in pneumonia include: Adenoviruses (in military recruits) [5] Metapneumovirus [7] Severe acute respiratory syndrome coronavirus (SARS-CoV) [8] Middle East respiratory syndrome virus (MERS-CoV) Hantaviruses [9] Viruses that primarily cause other diseases, but sometimes cause pneumonia include: Herpes simplex virus (HSV), mainly in newborns or young children Varicella-zoster virus (VZV) Measles virus Rubella virus Cytomegalovirus (CMV), mainly in people with immune system problems Smallpox virus Dengue virus The most commonly identified agents in children are respiratory syncytial virus , rhinovirus , human metapneumovirus , human bocavirus , and parainfluenza viruses . [7] History [ edit ] In the pre-antibiotic age, pneumonias had been treated with specific anti-serums of highly variable therapeutic effect and undesirable side-effects (a practice eliminated by the advent of sulfamides in 1936 and the beginning availability of penicillin in the 1940s). ... (October 2000). "Hantavirus Pulmonary Syndrome Is Distinguishable From Acute Interstitial Pneumonia" . ... PMID 21435708 . ^ a b c d e f g h Freeman, Andrew M.; Leigh, Jr (2020), "Viral Pneumonia" , StatPearls , Treasure Island (FL): StatPearls Publishing, PMID 30020658 , retrieved 2020-11-11 External links [ edit ] Classification D ICD - 10 : J12 ICD - 9-CM : 480 MeSH : D011024 SNOMED CT : 75570004 External resources MedlinePlus : 000073 eMedicine : emerg/468 radio/539 v t e Diseases of the respiratory system Upper RT (including URTIs , common cold ) Head sinuses Sinusitis nose Rhinitis Vasomotor rhinitis Atrophic rhinitis Hay fever Nasal polyp Rhinorrhea nasal septum Nasal septum deviation Nasal septum perforation Nasal septal hematoma tonsil Tonsillitis Adenoid hypertrophy Peritonsillar abscess Neck pharynx Pharyngitis Strep throat Laryngopharyngeal reflux (LPR) Retropharyngeal abscess larynx Croup Laryngomalacia Laryngeal cyst Laryngitis Laryngopharyngeal reflux (LPR) Laryngospasm vocal cords Laryngopharyngeal reflux (LPR) Vocal fold nodule Vocal fold paresis Vocal cord dysfunction epiglottis Epiglottitis trachea Tracheitis Laryngotracheal stenosis Lower RT / lung disease (including LRTIs ) Bronchial / obstructive acute Acute bronchitis chronic COPD Chronic bronchitis Acute exacerbation of COPD ) Asthma ( Status asthmaticus Aspirin-induced Exercise-induced Bronchiectasis Cystic fibrosis unspecified Bronchitis Bronchiolitis Bronchiolitis obliterans Diffuse panbronchiolitis Interstitial / restrictive ( fibrosis ) External agents/ occupational lung disease Pneumoconiosis Aluminosis Asbestosis Baritosis Bauxite fibrosis Berylliosis Caplan's syndrome Chalicosis Coalworker's pneumoconiosis Siderosis Silicosis Talcosis Byssinosis Hypersensitivity pneumonitis Bagassosis Bird fancier's lung Farmer's lung Lycoperdonosis Other ARDS Combined pulmonary fibrosis and emphysema Pulmonary edema Löffler's syndrome / Eosinophilic pneumonia Respiratory hypersensitivity Allergic bronchopulmonary aspergillosis Hamman-Rich syndrome Idiopathic pulmonary fibrosis Sarcoidosis Vaping-associated pulmonary injury Obstructive / Restrictive Pneumonia / pneumonitis By pathogen Viral Bacterial Pneumococcal Klebsiella Atypical bacterial Mycoplasma Legionnaires' disease Chlamydiae Fungal Pneumocystis Parasitic noninfectious Chemical / Mendelson's syndrome Aspiration / Lipid By vector/route Community-acquired Healthcare-associated Hospital-acquired By distribution Broncho- Lobar IIP UIP DIP BOOP-COP NSIP RB Other Atelectasis circulatory Pulmonary hypertension Pulmonary embolism Lung abscess Pleural cavity / mediastinum Pleural disease Pleuritis/pleurisy Pneumothorax / Hemopneumothorax Pleural effusion Hemothorax Hydrothorax Chylothorax Empyema/pyothorax Malignant Fibrothorax Mediastinal disease Mediastinitis Mediastinal emphysema Other/general Respiratory failure Influenza Common cold SARS Coronavirus disease 2019 Idiopathic pulmonary haemosiderosis Pulmonary alveolar proteinosis v t e Infectious diseases – viral systemic diseases Oncovirus DNA virus HBV Hepatocellular carcinoma HPV Cervical cancer Anal cancer Penile cancer Vulvar cancer Vaginal cancer Oropharyngeal cancer KSHV Kaposi's sarcoma EBV Nasopharyngeal carcinoma Burkitt's lymphoma Hodgkin lymphoma Follicular dendritic cell sarcoma Extranodal NK/T-cell lymphoma, nasal type MCPyV Merkel-cell carcinoma RNA virus HCV Hepatocellular carcinoma Splenic marginal zone lymphoma HTLV-I Adult T-cell leukemia/lymphoma Immune disorders HIV AIDS Central nervous system Encephalitis / meningitis DNA virus Human polyomavirus 2 Progressive multifocal leukoencephalopathy RNA virus MeV Subacute sclerosing panencephalitis LCV Lymphocytic choriomeningitis Arbovirus encephalitis Orthomyxoviridae (probable) Encephalitis lethargica RV Rabies Chandipura vesiculovirus Herpesviral meningitis Ramsay Hunt syndrome type 2 Myelitis Poliovirus Poliomyelitis Post-polio syndrome HTLV-I Tropical spastic paraparesis Eye Cytomegalovirus Cytomegalovirus retinitis HSV Herpes of the eye Cardiovascular CBV Pericarditis Myocarditis Respiratory system / acute viral nasopharyngitis / viral pneumonia DNA virus Epstein–Barr virus EBV infection / Infectious mononucleosis Cytomegalovirus RNA virus IV : Human coronavirus 229E / NL63 / HKU1 / OC43 Common cold MERS coronavirus Middle East respiratory syndrome SARS coronavirus Severe acute respiratory syndrome SARS coronavirus 2 Coronavirus disease 2019 V , Orthomyxoviridae : Influenza virus A / B / C / D Influenza / Avian influenza V, Paramyxoviridae : Human parainfluenza viruses Parainfluenza Human orthopneumovirus hMPV Human digestive system Pharynx / Esophagus MuV Mumps Cytomegalovirus Cytomegalovirus esophagitis Gastroenteritis / diarrhea DNA virus Adenovirus Adenovirus infection RNA virus Rotavirus Norovirus Astrovirus Coronavirus Hepatitis DNA virus HBV ( B ) RNA virus CBV HAV ( A ) HCV ( C ) HDV ( D ) HEV ( E ) HGV ( G ) Pancreatitis CBV Urogenital BK virus MuV Mumps v t e Pneumonia Infectious pneumonias Bacterial pneumonia Viral pneumonia Fungal pneumonia Parasitic pneumonia Atypical pneumonia Community-acquired pneumonia Healthcare-associated pneumonia Hospital-acquired pneumonia Ventilator-associated pneumonia Severe acute respiratory syndrome Pneumonias caused by infectious or noninfectious agents Aspiration pneumonia Lipid pneumonia Eosinophilic pneumonia Bronchiolitis obliterans organizing pneumonia Noninfectious pneumonia Chemical pneumonitis Idiopathic pneumonia syndrome