-
Loeffler Endocarditis
Wikipedia
This stage is dominated by signs and symptoms of the acute coronary syndrome such as angina , heart attack , and congestive heart failure . ... For example, eosinophilic granulomatosis with polyangiitis can be successfully treated with mepolizumab . Idiopathic hypereosinphilic syndrome and lymphocyte-variant hypereosinophilia: corticosteroids; for individuals with these hypereosinophilias that are refractory to or break through corticosteroid therapy and individuals requiring corticosteroid-sparing therapy, recommended alternative drug therapies include hydroxyurea , Pegylated interferon -α, and either one of the tyrosine kinase inhibitors imatinib and mepolizumab). ... PMID 29044676 . ^ Kleinfeldt, Tilo; Ince, Hueseyin; Nienaber, Christoph A. (2011). "Hypereosinophilic Syndrome: A rare case of Loeffler's endocarditis documented in cardiac MRI". ... "Management of Hypereosinophilic Syndromes". Immunology and Allergy Clinics of North America . 35 (3): 561–75. doi : 10.1016/j.iac.2015.05.006 . ... External links [ edit ] Classification D ICD - 10 : I42.3 ICD - 9-CM : 421.0 MeSH : D017681 DiseasesDB : 4291 External resources eMedicine : med/1318 Orphanet : 75566 v t e Cardiovascular disease (heart) Ischaemic Coronary disease Coronary artery disease (CAD) Coronary artery aneurysm Spontaneous coronary artery dissection (SCAD) Coronary thrombosis Coronary vasospasm Myocardial bridge Active ischemia Angina pectoris Prinzmetal's angina Stable angina Acute coronary syndrome Myocardial infarction Unstable angina Sequelae hours Hibernating myocardium Myocardial stunning days Myocardial rupture weeks Aneurysm of heart / Ventricular aneurysm Dressler syndrome Layers Pericardium Pericarditis Acute Chronic / Constrictive Pericardial effusion Cardiac tamponade Hemopericardium Myocardium Myocarditis Chagas disease Cardiomyopathy Dilated Alcoholic Hypertrophic Tachycardia-induced Restrictive Loeffler endocarditis Cardiac amyloidosis Endocardial fibroelastosis Arrhythmogenic right ventricular dysplasia Endocardium / valves Endocarditis infective endocarditis Subacute bacterial endocarditis non-infective endocarditis Libman–Sacks endocarditis Nonbacterial thrombotic endocarditis Valves mitral regurgitation prolapse stenosis aortic stenosis insufficiency tricuspid stenosis insufficiency pulmonary stenosis insufficiency Conduction / arrhythmia Bradycardia Sinus bradycardia Sick sinus syndrome Heart block : Sinoatrial AV 1° 2° 3° Intraventricular Bundle branch block Right Left Left anterior fascicle Left posterior fascicle Bifascicular Trifascicular Adams–Stokes syndrome Tachycardia ( paroxysmal and sinus ) Supraventricular Atrial Multifocal Junctional AV nodal reentrant Junctional ectopic Ventricular Accelerated idioventricular rhythm Catecholaminergic polymorphic Torsades de pointes Premature contraction Atrial Junctional Ventricular Pre-excitation syndrome Lown–Ganong–Levine Wolff–Parkinson–White Flutter / fibrillation Atrial flutter Ventricular flutter Atrial fibrillation Familial Ventricular fibrillation Pacemaker Ectopic pacemaker / Ectopic beat Multifocal atrial tachycardia Pacemaker syndrome Parasystole Wandering atrial pacemaker Long QT syndrome Andersen–Tawil Jervell and Lange-Nielsen Romano–Ward Cardiac arrest Sudden cardiac death Asystole Pulseless electrical activity Sinoatrial arrest Other / ungrouped hexaxial reference system Right axis deviation Left axis deviation QT Short QT syndrome T T wave alternans ST Osborn wave ST elevation ST depression Strain pattern Cardiomegaly Ventricular hypertrophy Left Right / Cor pulmonale Atrial enlargement Left Right Athletic heart syndrome Other Cardiac fibrosis Heart failure Diastolic heart failure Cardiac asthma Rheumatic fever
-
Wfs1 Wolfram Syndrome Spectrum Disorder
Gene_reviews
Note: This GeneReview focuses on Wolfram syndrome spectrum disorder caused by biallelic WFS1 pathogenic variants. ... SNHL in 13% of individuals Slowly progressive ataxia w/mean onset age 10-15 yrs (usually <25 yrs); dysarthria, muscle weakness, spasticity in the lower limbs, scoliosis, bladder dysfunction, absent lower limb reflexes, & loss of position & vibration sense mtDNA deletion Kearns-Sayre syndrome (See Mitochondrial DNA Deletion Syndromes.) ... Listed genes represent the most commonly associated genes; at least 19 genes are associated with Bardet-Biedl syndrome (see Bardet-Biedl Syndrome). Optic atrophy associated with hearing impairment. ... See also Wolfram Syndrome Clinical Management Guidelines, page 5 for recommended baseline investigations. ... OT = occupational therapy; PT = physical therapy 1. For details see Wolfram Syndrome Clinical Management Guidelines, pages 6-12.
-
Acute Radiation Syndrome
Wikipedia
Each syndrome requires that the tissue showing the syndrome itself be exposed (e.g., gastrointestinal syndrome is not seen if the stomach and intestines are not exposed to radiation). Some areas affected are: Hematopoietic. This syndrome is marked by a drop in the number of blood cells , called aplastic anemia . ... "chronic%20radiation%20syndrome"&pg=PA1 Chronic Radiation Syndrome . Springer Science & Business Media. p. 1. ... PMID 15080238 . ^ "Time Phases of Acute Radiation Syndrome (ARS) - Dose >8 Gy" . Radiation Emergency Medical Management . ... Centers for Disease Control and Prevention External links [ edit ] Nuclear technology portal Wikimedia Commons has media related to Acute radiation syndrome . "Fact sheet on Acute Radiation Syndrome" .

-
Epilepsy-Intellectual Disability In Females
Wikipedia
PCDH19 gene-related epilepsy is thought to develop based upon a deficiency of the calcium-dependent cell-adhesion PCDH19 (protocadherin 19) gene. [24] [nb 1] Its cause and pathophysiology (cause and mechanisms by which damage occurs) are different from other epilepsies, although the symptoms are very similar to other epileptic syndromes, such as Generalized epilepsy with febrile seizures plus (GEFS+), Dravet syndrome with SCN1A negative, FIRES (febrile infection–related epilepsy syndrome) Lennox-Gastaut syndrome or epilepsy of unknown origin. [25] Diagnosis [ edit ] PCDH19 gene-related epilepsy is clinically based on patient and family seizure history, cognitive and behavioral neuropsychological evaluation, neurological examination, electroencephalogram (EEG) studies, and long term observation. ... "Acute-onset epilepsy triggered by fever mimicking FIRES febrile infection–related epilepsy syndrome: The role of protocadherin 19 (PCDH19) gene mutation". ... CS1 maint: numeric names: authors list ( link ) ^ a b "PCDH19 and SCN8A Research Funded By The Cute Syndrome" . The Cute Syndrome Foundation: Funding PCDH19 Epilepsy & SCN8A Epilepsy Research . ... Retrieved 2016-08-22 . ^ a b c d e f g "News about PCDH19 Epilepsy and SCN8A Epilepsy and The Cute Syndrome Foundation" . The Cute Syndrome Foundation: Funding PCDH19 Epilepsy & SCN8A Epilepsy Research . ... "Reversal of Neurological Defects in a Mouse Model of Rett Syndrome". Science . 315 (5815): 1143–1147.
-
Neonatal Conjunctivitis
Wikipedia
External links [ edit ] Classification D ICD - 10 : A54.3 , P39.1 ICD - 9-CM : 098.40 , 771.6 MeSH : D009878 DiseasesDB : 9237 External resources MedlinePlus : 001606 eMedicine : oph/325 v t e Diseases of the human eye Adnexa Eyelid Inflammation Stye Chalazion Blepharitis Entropion Ectropion Lagophthalmos Blepharochalasis Ptosis Blepharophimosis Xanthelasma Ankyloblepharon Eyelash Trichiasis Madarosis Lacrimal apparatus Dacryoadenitis Epiphora Dacryocystitis Xerophthalmia Orbit Exophthalmos Enophthalmos Orbital cellulitis Orbital lymphoma Periorbital cellulitis Conjunctiva Conjunctivitis allergic Pterygium Pseudopterygium Pinguecula Subconjunctival hemorrhage Globe Fibrous tunic Sclera Scleritis Episcleritis Cornea Keratitis herpetic acanthamoebic fungal Exposure Photokeratitis Corneal ulcer Thygeson's superficial punctate keratopathy Corneal dystrophy Fuchs' Meesmann Corneal ectasia Keratoconus Pellucid marginal degeneration Keratoglobus Terrien's marginal degeneration Post-LASIK ectasia Keratoconjunctivitis sicca Corneal opacity Corneal neovascularization Kayser–Fleischer ring Haab's striae Arcus senilis Band keratopathy Vascular tunic Iris Ciliary body Uveitis Intermediate uveitis Hyphema Rubeosis iridis Persistent pupillary membrane Iridodialysis Synechia Choroid Choroideremia Choroiditis Chorioretinitis Lens Cataract Congenital cataract Childhood cataract Aphakia Ectopia lentis Retina Retinitis Chorioretinitis Cytomegalovirus retinitis Retinal detachment Retinoschisis Ocular ischemic syndrome / Central retinal vein occlusion Central retinal artery occlusion Branch retinal artery occlusion Retinopathy diabetic hypertensive Purtscher's of prematurity Bietti's crystalline dystrophy Coats' disease Sickle cell Macular degeneration Retinitis pigmentosa Retinal haemorrhage Central serous retinopathy Macular edema Epiretinal membrane (Macular pucker) Vitelliform macular dystrophy Leber's congenital amaurosis Birdshot chorioretinopathy Other Glaucoma / Ocular hypertension / Primary juvenile glaucoma Floater Leber's hereditary optic neuropathy Red eye Globe rupture Keratomycosis Phthisis bulbi Persistent fetal vasculature / Persistent hyperplastic primary vitreous Persistent tunica vasculosa lentis Familial exudative vitreoretinopathy Pathways Optic nerve Optic disc Optic neuritis optic papillitis Papilledema Foster Kennedy syndrome Optic atrophy Optic disc drusen Optic neuropathy Ischemic anterior (AION) posterior (PION) Kjer's Leber's hereditary Toxic and nutritional Strabismus Extraocular muscles Binocular vision Accommodation Paralytic strabismus Ophthalmoparesis Chronic progressive external ophthalmoplegia Kearns–Sayre syndrome palsies Oculomotor (III) Fourth-nerve (IV) Sixth-nerve (VI) Other strabismus Esotropia / Exotropia Hypertropia Heterophoria Esophoria Exophoria Cyclotropia Brown's syndrome Duane syndrome Other binocular Conjugate gaze palsy Convergence insufficiency Internuclear ophthalmoplegia One and a half syndrome Refraction Refractive error Hyperopia Myopia Astigmatism Anisometropia / Aniseikonia Presbyopia Vision disorders Blindness Amblyopia Leber's congenital amaurosis Diplopia Scotoma Color blindness Achromatopsia Dichromacy Monochromacy Nyctalopia Oguchi disease Blindness / Vision loss / Visual impairment Anopsia Hemianopsia binasal bitemporal homonymous Quadrantanopia subjective Asthenopia Hemeralopia Photophobia Scintillating scotoma Pupil Anisocoria Argyll Robertson pupil Marcus Gunn pupil Adie syndrome Miosis Mydriasis Cycloplegia Parinaud's syndrome Other Nystagmus Childhood blindness Infections Trachoma Onchocerciasis v t e Conditions originating in the perinatal period / fetal disease Maternal factors complicating pregnancy, labour or delivery placenta Placenta praevia Placental insufficiency Twin-to-twin transfusion syndrome chorion / amnion Chorioamnionitis umbilical cord Umbilical cord prolapse Nuchal cord Single umbilical artery presentation Breech birth Asynclitism Shoulder presentation Growth Small for gestational age / Large for gestational age Preterm birth / Postterm pregnancy Intrauterine growth restriction Birth trauma scalp Cephalohematoma Chignon Caput succedaneum Subgaleal hemorrhage Brachial plexus injury Erb's palsy Klumpke paralysis Affected systems Respiratory Intrauterine hypoxia Infant respiratory distress syndrome Transient tachypnea of the newborn Meconium aspiration syndrome Pleural disease Pneumothorax Pneumomediastinum Wilson–Mikity syndrome Bronchopulmonary dysplasia Cardiovascular Pneumopericardium Persistent fetal circulation Bleeding and hematologic disease Vitamin K deficiency bleeding HDN ABO Anti-Kell Rh c Rh D Rh E Hydrops fetalis Hyperbilirubinemia Kernicterus Neonatal jaundice Velamentous cord insertion Intraventricular hemorrhage Germinal matrix hemorrhage Anemia of prematurity Gastrointestinal Ileus Necrotizing enterocolitis Meconium peritonitis Integument and thermoregulation Erythema toxicum Sclerema neonatorum Nervous system Perinatal asphyxia Periventricular leukomalacia Musculoskeletal Gray baby syndrome muscle tone Congenital hypertonia Congenital hypotonia Infections Vertically transmitted infection Neonatal infection rubella herpes simplex mycoplasma hominis ureaplasma urealyticum Omphalitis Neonatal sepsis Group B streptococcal infection Neonatal conjunctivitis Other Miscarriage Perinatal mortality Stillbirth Infant mortality Neonatal withdrawal

-
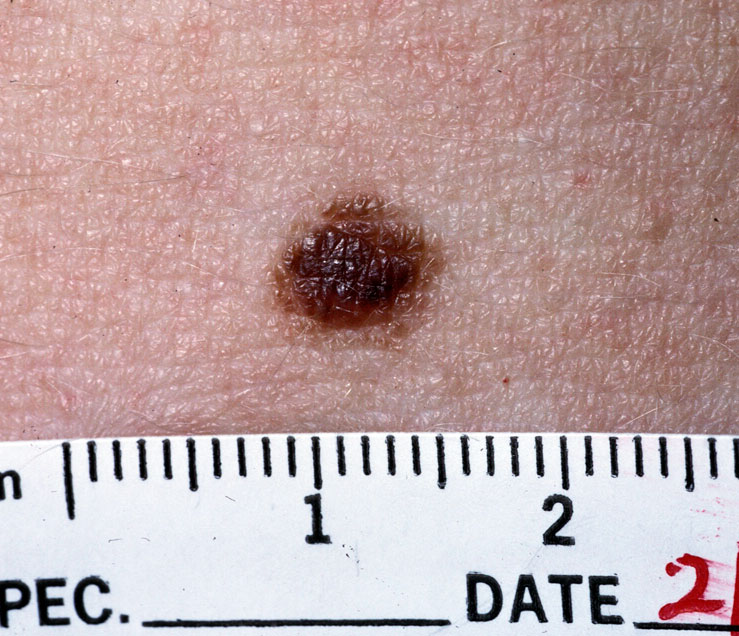
Nevus
Wikipedia
Contents 1 Classification 1.1 Increased melanin 1.1.1 Usually acquired 1.1.2 Usually congenital 1.2 Decreased melanin 1.2.1 Acquired 1.2.2 Congenital 1.3 Epidermal nevi 1.4 Connective tissue nevi 1.5 Vascular nevi 1.6 Intramucosal nevi 2 Diagnosis 2.1 Differential diagnoses 3 Management 3.1 Observation 3.2 Destruction 3.3 Surgery 4 Syndromes 5 Etymology 6 References 7 External links Classification [ edit ] The term nevus is applied to a number of conditions caused by neoplasias and hyperplasias of melanocytes , [2] as well as a number of pigmentation disorders, both hypermelanotic (containing increased melanin , the pigment responsible for skin color) and hypomelanotic (containing decreased melanin). [3] Skin lesions which are multi-colored or polychromatic may be a finding in skin cancer . [4] Increased melanin [ edit ] Usually acquired [ edit ] Melanocytic nevus Melanocytic nevi can be categorized based on the location of melanocytic cells [2] Junctional: epidermis Intradermal: dermis Compound: epidermis and dermis Atypical (dysplastic) nevus : This type of nevus must be diagnosed based on histological features. ... A modern polarized dermatoscope A dermatoscope Differential diagnoses [ edit ] Hypermelanotic nevi must be differentiated from other types of pigmented skin lesions, including: [6] [7] Lentigo simplex Solar lentigo Café au lait macule Ink-spot lentigo Mucosal melanotic macule Mongolian spot (dermal melanocytosis) Cafe au lait Mongolian spot Management [ edit ] Cryotherapy The management of a nevus depends on the specific diagnosis, however, the options for treatment generally include the following modalities: Observation [ edit ] Destruction [ edit ] Chemical peels Cryotherapy Dermabrasion Electrodessication Laser ablation Surgery [ edit ] The decision to observe or treat a nevus may depend on a number of factors, including cosmetic concerns, irritative symptoms (e.g., pruritus), ulceration, infection, and concern for potential malignancy. [2] Syndromes [ edit ] The term nevus is included in the names of multiple dermatologic syndromes: Basal cell nevus syndrome Blue rubber bleb nevus syndrome Dysplastic nevus syndrome Epidermal nevus syndrome Linear nevus sebaceous syndrome Etymology [ edit ] A nevus may also be spelled naevus. ... Wikimedia Commons has media related to Nevus . v t e Diseases of the skin and appendages by morphology Growths Epidermal Wart Callus Seborrheic keratosis Acrochordon Molluscum contagiosum Actinic keratosis Squamous-cell carcinoma Basal-cell carcinoma Merkel-cell carcinoma Nevus sebaceous Trichoepithelioma Pigmented Freckles Lentigo Melasma Nevus Melanoma Dermal and subcutaneous Epidermal inclusion cyst Hemangioma Dermatofibroma (benign fibrous histiocytoma) Keloid Lipoma Neurofibroma Xanthoma Kaposi's sarcoma Infantile digital fibromatosis Granular cell tumor Leiomyoma Lymphangioma circumscriptum Myxoid cyst Rashes With epidermal involvement Eczematous Contact dermatitis Atopic dermatitis Seborrheic dermatitis Stasis dermatitis Lichen simplex chronicus Darier's disease Glucagonoma syndrome Langerhans cell histiocytosis Lichen sclerosus Pemphigus foliaceus Wiskott–Aldrich syndrome Zinc deficiency Scaling Psoriasis Tinea ( Corporis Cruris Pedis Manuum Faciei ) Pityriasis rosea Secondary syphilis Mycosis fungoides Systemic lupus erythematosus Pityriasis rubra pilaris Parapsoriasis Ichthyosis Blistering Herpes simplex Herpes zoster Varicella Bullous impetigo Acute contact dermatitis Pemphigus vulgaris Bullous pemphigoid Dermatitis herpetiformis Porphyria cutanea tarda Epidermolysis bullosa simplex Papular Scabies Insect bite reactions Lichen planus Miliaria Keratosis pilaris Lichen spinulosus Transient acantholytic dermatosis Lichen nitidus Pityriasis lichenoides et varioliformis acuta Pustular Acne vulgaris Acne rosacea Folliculitis Impetigo Candidiasis Gonococcemia Dermatophyte Coccidioidomycosis Subcorneal pustular dermatosis Hypopigmented Tinea versicolor Vitiligo Pityriasis alba Postinflammatory hyperpigmentation Tuberous sclerosis Idiopathic guttate hypomelanosis Leprosy Hypopigmented mycosis fungoides Without epidermal involvement Red Blanchable Erythema Generalized Drug eruptions Viral exanthems Toxic erythema Systemic lupus erythematosus Localized Cellulitis Abscess Boil Erythema nodosum Carcinoid syndrome Fixed drug eruption Specialized Urticaria Erythema ( Multiforme Migrans Gyratum repens Annulare centrifugum Ab igne ) Nonblanchable Purpura Macular Thrombocytopenic purpura Actinic/solar purpura Papular Disseminated intravascular coagulation Vasculitis Indurated Scleroderma / morphea Granuloma annulare Lichen sclerosis et atrophicus Necrobiosis lipoidica Miscellaneous disorders Ulcers Hair Telogen effluvium Androgenic alopecia Alopecia areata Systemic lupus erythematosus Tinea capitis Loose anagen syndrome Lichen planopilaris Folliculitis decalvans Acne keloidalis nuchae Nail Onychomycosis Psoriasis Paronychia Ingrown nail Mucous membrane Aphthous stomatitis Oral candidiasis Lichen planus Leukoplakia Pemphigus vulgaris Mucous membrane pemphigoid Cicatricial pemphigoid Herpesvirus Coxsackievirus Syphilis Systemic histoplasmosis Squamous-cell carcinoma v t e Skin cancer of nevi and melanomas Melanoma Mucosal melanoma Superficial spreading melanoma Nodular melanoma lentigo Lentigo maligna / Lentigo maligna melanoma Acral lentiginous melanoma Amelanotic melanoma Desmoplastic melanoma Melanoma with features of a Spitz nevus Melanoma with small nevus-like cells Polypoid melanoma Nevoid melanoma Melanocytic tumors of uncertain malignant potential Nevus / melanocytic nevus Nevus of Ito / Nevus of Ota Spitz nevus Pigmented spindle cell nevus Halo nevus Pseudomelanoma Blue nevus of Jadassohn–Tièche Cellular Epithelioid Deep penetrating Amelanotic Malignant Congenital melanocytic nevus ( Giant Medium-sized Small-sized ) Balloon cell nevus Dysplastic nevus / Dysplastic nevus syndrome Acral nevus Becker's nevus Benign melanocytic nevus Nevus spilusPLA2G6, MTAP, MC1R, CDKN2A, BAP1, TERT, CDKN2B, NID1, HRAS, MITF, CDK4, PTEN, POT1, MYH3, H1-4, TRIM37, PIK3CA, PRKAR1A, PTPN11, SOS1, MGMT, AKT1, TP63, MED25, CDC42, MAFF, TERF2IP, ASXL2, HDAC8, ACD, ATP5F1D, CHRNG, CYP26C1, BRAF, NRAS, TP53, IRF4, MLANA, KHDC3L, HSDL1, NLRP7, BCAR1, KITLG, TMX2-CTNND1, KIT, H3P10, EDN1, CTNND1, CSE1L, FGFR3, IL1B, SYNE1, ERVW-1, NXT1, DDIT3, NQO1, PRAME, CARD8, IL23A, SUB1, MRPL28, CLOCK, DMBT1, HSPB3, DNM1, DSG2, NSDHL, GADD45A, XRCC3, KRT20, H3P9, ALCAM, AD12, MIR373, RTL1, BAD, BCL2, SASS6, LINC00032, MS4A1, CDK6, CDKN1A, AMN, CLPTM1L, CTNNB1, COIL, ELAVL2, IL10, CITED1, NOS2, GNAQ, NGFR, HCCS, MYC, HCL2, HSPB1, HSPB2, TLR4, IFN1@, IFNA1, IFNA2, LRP2, IFNA13, IFNA17, IL18, GJB2, OCA2, PAX3, PAX4, ERBB2, ETS1, SUMO3, RHCE, RAG2, RAF1, EZH2, FGF2, PTCH1, HTRA1, PPARG, PKD1, GALK1, PCNA, PAX9, PARP1
-
Atelectasis
Wikipedia
In premature babies , this leads to infant respiratory distress syndrome . The term uses combining forms of atel- + ectasis , from Greek : ἀτελής , "incomplete" + ἔκτασις, "extension". ... Leakage of air into the pleural cavity ( pneumothorax ) also leads to compression atelectasis. [11] Cicatrization (contraction) atelectasis [ edit ] It occurs when either local or generalized fibrotic changes in the lung or pleura hamper expansion and increase elastic recoil during expiration. [11] Causes include granulomatous disease, necrotising pneumonia and radiation fibrosis. [12] Chronic atelectasis [ edit ] Chronic atelectasis may take one of two forms—middle lobe syndrome or rounded atelectasis. Right middle lobe syndrome [ edit ] In right middle lobe syndrome, the middle lobe of the right lung contracts, usually because of pressure on the bronchus from enlarged lymph glands and occasionally a tumor . ... Patchy atelectasis [ edit ] Is due to lack of surfactant, as occurs in hyaline membrane disease of newborn or acute (adult) respiratory distress syndrome (ARDS). [13] Rounded atelectasis [ edit ] In rounded atelectasis (folded lung or Blesovsky syndrome [14] ), an outer portion of the lung slowly collapses as a result of scarring and shrinkage of the membrane layers covering the lungs ( pleura ), which would show as visceral pleural thickening and entrapment of lung tissue. ... See also [ edit ] Alveolar capillary dysplasia , a very rare type of diffuse congenital disorder of the lung Flat-chested kitten syndrome or FCKS: atelectasis in neo-natal kittens Tympanic membrane atelectasis : Retraction of the ear drum into the middle ear can also be referred to as atelectasis. ... "Lung folding simulating peripheral pulmonary neoplasm (Blesovsky's syndrome)" . Thorax . 35 (12): 936–940. doi : 10.1136/thx.35.12.936 .ARVCF, CCDC114, DNAAF5, CCDC40, ARMC4, DNAAF2, CFAP298, DNAI2, TTC25, DNAL1, RSPH3, CFAP300, CCDC65, RSPH1, DRC1, LRRC56, COMT, CCDC151, DNAAF1, PIH1D3, DNAAF4, JMJD1C, RSPH9, GAS2L2, CCDC39, MCIDAS, RSPH4A, DNAAF3, NHLRC2, DNAJB13, HYDIN, RIPK4, ZMYND10, NME8, DNAH5, GAS8, GP1BB, LAMA2, OCRL, PAX3, PLEC, RPGR, RREB1, SPAG1, STAT3, TBX1, TSC1, TSC2, HIRA, UFD1, CXCR4, OFD1, DNAH11, SEC24C, CCNO, FBLN5, LRRC6, DNAH1, DNAI1, STK36, EFEMP2, CCDC103

-
Pseudoachondroplasia
Wikipedia
Pseudoachondroplasia is usually first detected by a drop of linear growth in contrast to peers, a waddling gait or arising lower limb deformities. [2] Pseudoachondroplasia (also known as PSACH, pseudoachondroplastic dysplasia, and pseudoachondroplastic spondyloepiphyseal dysplasia syndrome) is an osteochondrodysplasia that results in mild to severely short stature due to the inhibition of skeletal growth primarily in the limbs. [2] Though similarities in nomenclature may cause confusion, pseudoachondroplasia should not be confused with achondroplasia , which is a clinically and genetically distinct skeletal dysplasia . ... “Pseudoachondroplasia, PSACH” v t e Diseases of collagen , laminin and other scleroproteins Collagen disease COL1 : Osteogenesis imperfecta Ehlers–Danlos syndrome, types 1, 2, 7 COL2 : Hypochondrogenesis Achondrogenesis type 2 Stickler syndrome Marshall syndrome Spondyloepiphyseal dysplasia congenita Spondyloepimetaphyseal dysplasia, Strudwick type Kniest dysplasia (see also C2/11 ) COL3 : Ehlers–Danlos syndrome, types 3 & 4 Sack–Barabas syndrome COL4 : Alport syndrome COL5 : Ehlers–Danlos syndrome, types 1 & 2 COL6 : Bethlem myopathy Ullrich congenital muscular dystrophy COL7 : Epidermolysis bullosa dystrophica Recessive dystrophic epidermolysis bullosa Bart syndrome Transient bullous dermolysis of the newborn COL8: Fuchs' dystrophy 1 COL9: Multiple epiphyseal dysplasia 2, 3, 6 COL10: Schmid metaphyseal chondrodysplasia COL11: Weissenbacher–Zweymüller syndrome Otospondylomegaepiphyseal dysplasia (see also C2/11 ) COL17: Bullous pemphigoid COL18: Knobloch syndrome Laminin Junctional epidermolysis bullosa Laryngoonychocutaneous syndrome Other Congenital stromal corneal dystrophy Raine syndrome Urbach–Wiethe disease TECTA DFNA8/12, DFNB21 see also fibrous proteinsCOMP, COL9A2, COL9A3, COL9A1, SCN8A, MATN3, ACAN, FGFR3, SPARC, P4HB, GLS, DCN, EPHA3, DMD, CANX, HAPLN1, COL10A1, COL2A1, MMRN1

-
Femoral Hernia
Wikipedia
External links [ edit ] Classification D ICD - 10 : K41 ICD - 9-CM : 553.0 MeSH : D006550 DiseasesDB : 4793 External resources MedlinePlus : 001136 eMedicine : emerg/251 Surgery Encyclopaedia v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum
-
Multiple Endocrine Neoplasia, Type Iib
Omim
The features suggesting Marfan syndrome were high arched palate, pectus excavatum, bilateral pes cavus, high patella and scoliosis. ... Morrison and Nevin (1996) presented a review of this syndrome. They used the term Wagenmann-Froboese syndrome, because Wagenmann (1922) and Froboese (1923) initially described this disorder. ... Gordon et al. (1998) concluded that pure mucosal neuroma syndrome does not appear to be a forme fruste of MEN2B at the genetic level. ... Leboulleux et al. (2002) concluded that the study confirmed the need for early treatment of MTC in patients with the MEN2B syndrome, preferably within the first 6 months of life. The phenotype of MTC occurring in the MEN2B syndrome was, however, not more aggressive than sporadic MTC or MTC occurring in other familial syndromes.RET, CALCA, MEN1, RBP3, SDHD, NTRK1, MT1A, GDNF, ITGB1, VHL, HNRNPA3P1, HNRNPA3, PNMT, SLC16A1, SDHB, MCAT, MCPH1, AR, TGM2, S100A12, RYK, SDHC, SLC6A2, ABO, SLC18A1, SLC18A2, PTEN, TP53, VEGFA, VGF, ZNF22, NCOA4, OSMR, SIN3A, SESN2, IL31RA, MIR885, TH, NPY, PRL, POMC, BRCA1, BRCA2, CEACAM5, CFTR, CGA, CHGA, DBH, ACE, EGR1, EIF4G1, EPAS1, EPO, ESR1, FH, GC, GCG, GPT, NR3C1, HLA-H, HNRNPF, HP, MAZ, MBL2, MT2A, MT2P1, NF1, AKT1, MIR1225
-
Kjer's Optic Neuropathy
Wikipedia
Contents 1 Presentation 2 Genetics 3 Pathophysiology 4 Management 5 Incidence 6 See also 7 References 8 Further reading 9 External links Presentation [ edit ] Autosomal dominant optic atrophy can present clinically as an isolated bilateral optic neuropathy (non-syndromic form) or rather as a complicated phenotype with extra-ocular signs (syndromic form). ... External links [ edit ] Classification D ICD - 9-CM : 377.16 OMIM : 165500 MeSH : D029241 DiseasesDB : 34452 v t e Diseases of the human eye Adnexa Eyelid Inflammation Stye Chalazion Blepharitis Entropion Ectropion Lagophthalmos Blepharochalasis Ptosis Blepharophimosis Xanthelasma Ankyloblepharon Eyelash Trichiasis Madarosis Lacrimal apparatus Dacryoadenitis Epiphora Dacryocystitis Xerophthalmia Orbit Exophthalmos Enophthalmos Orbital cellulitis Orbital lymphoma Periorbital cellulitis Conjunctiva Conjunctivitis allergic Pterygium Pseudopterygium Pinguecula Subconjunctival hemorrhage Globe Fibrous tunic Sclera Scleritis Episcleritis Cornea Keratitis herpetic acanthamoebic fungal Exposure Photokeratitis Corneal ulcer Thygeson's superficial punctate keratopathy Corneal dystrophy Fuchs' Meesmann Corneal ectasia Keratoconus Pellucid marginal degeneration Keratoglobus Terrien's marginal degeneration Post-LASIK ectasia Keratoconjunctivitis sicca Corneal opacity Corneal neovascularization Kayser–Fleischer ring Haab's striae Arcus senilis Band keratopathy Vascular tunic Iris Ciliary body Uveitis Intermediate uveitis Hyphema Rubeosis iridis Persistent pupillary membrane Iridodialysis Synechia Choroid Choroideremia Choroiditis Chorioretinitis Lens Cataract Congenital cataract Childhood cataract Aphakia Ectopia lentis Retina Retinitis Chorioretinitis Cytomegalovirus retinitis Retinal detachment Retinoschisis Ocular ischemic syndrome / Central retinal vein occlusion Central retinal artery occlusion Branch retinal artery occlusion Retinopathy diabetic hypertensive Purtscher's of prematurity Bietti's crystalline dystrophy Coats' disease Sickle cell Macular degeneration Retinitis pigmentosa Retinal haemorrhage Central serous retinopathy Macular edema Epiretinal membrane (Macular pucker) Vitelliform macular dystrophy Leber's congenital amaurosis Birdshot chorioretinopathy Other Glaucoma / Ocular hypertension / Primary juvenile glaucoma Floater Leber's hereditary optic neuropathy Red eye Globe rupture Keratomycosis Phthisis bulbi Persistent fetal vasculature / Persistent hyperplastic primary vitreous Persistent tunica vasculosa lentis Familial exudative vitreoretinopathy Pathways Optic nerve Optic disc Optic neuritis optic papillitis Papilledema Foster Kennedy syndrome Optic atrophy Optic disc drusen Optic neuropathy Ischemic anterior (AION) posterior (PION) Kjer's Leber's hereditary Toxic and nutritional Strabismus Extraocular muscles Binocular vision Accommodation Paralytic strabismus Ophthalmoparesis Chronic progressive external ophthalmoplegia Kearns–Sayre syndrome palsies Oculomotor (III) Fourth-nerve (IV) Sixth-nerve (VI) Other strabismus Esotropia / Exotropia Hypertropia Heterophoria Esophoria Exophoria Cyclotropia Brown's syndrome Duane syndrome Other binocular Conjugate gaze palsy Convergence insufficiency Internuclear ophthalmoplegia One and a half syndrome Refraction Refractive error Hyperopia Myopia Astigmatism Anisometropia / Aniseikonia Presbyopia Vision disorders Blindness Amblyopia Leber's congenital amaurosis Diplopia Scotoma Color blindness Achromatopsia Dichromacy Monochromacy Nyctalopia Oguchi disease Blindness / Vision loss / Visual impairment Anopsia Hemianopsia binasal bitemporal homonymous Quadrantanopia subjective Asthenopia Hemeralopia Photophobia Scintillating scotoma Pupil Anisocoria Argyll Robertson pupil Marcus Gunn pupil Adie syndrome Miosis Mydriasis Cycloplegia Parinaud's syndrome Other Nystagmus Childhood blindness Infections Trachoma Onchocerciasis v t e Mitochondrial diseases Carbohydrate metabolism PCD PDHA Primarily nervous system Leigh disease LHON NARP Myopathies KSS Mitochondrial encephalomyopathy MELAS MERRF PEO No primary system DAD MNGIE Pearson syndrome Chromosomal OPA1 Kjer's optic neuropathy SARS2 HUPRA syndrome TIMM8A Mohr–Tranebjærg syndrome see also mitochondrial proteinsOPA1, DNM1L, OPA3, MFN2, MFN1, DENR, DAPK2, OMA1, FIS1, UTRN, CRMP1, SIRT3, IMMT, MFF, MED12, YME1L1, SSBP1, PRKAB1, NFE2L2, PPARGC1A, PPARG, PRKAA1, KIF1B, OPA5, PRKAA2, GSTM1, FXN, WFS1, SLC14A1, LEP, TOMM70, TOMM40, SQSTM1, YAP1, OPTN, COX5A, MRPS30, MAD2L1BP, KEAP1, PAPOLA, ACO2, TPPP, PACC1, PGAM5, CCDC50, MCU, MAP1LC3B, MUL1, PINK1, TFB2M, OPA4, SEMA6A, ADCK1, MTPAP, TREH, APTX, TMED9, ASAP1, CD274, FGF21, GABARAPL1, TUSC2, CDK5R1, PDAP1, PHB2, ASAP2, TP53BP1, BECN1, CNR2, GABPA, ERG, EPHB2, ELAVL1, DGKG, CYP1B1, CYC1, CTRL, CPOX, COX8A, CDKN1A, AKAP1, CDH1, CAV1, CAST, CAPN1, BNIP3L, BNIP3, CEACAM1, BGLAP, BCL2, AFM, GSTT1, H2AX, HRES1, HES1, VEGFA, PHLDA2, ACP3, CLDN5, SPG7, SOD1, SLC5A1, SDHA, MAPK1, PRKCA, PLXNA2, PLIN1, PKLR, REG3A, NRF1, NOS3, NFE2L1, NDUFV2, MYC, LCN2, LAMP1, OPA8
-
Hemiparesis
Wikipedia
Pure Motor Hemiparesis, a form of hemiparesis characterized by sided weakness in the leg, arm, and face, is the most commonly diagnosed form of hemiparesis. [1] Pusher syndrome [ edit ] Main article: Pusher syndrome Pusher syndrome is a clinical disorder following left or right brain damage in which patients actively push their weight away from the nonhemiparetic side to the hemiparetic side. ... The increased risk of falls must be addressed with therapy to correct their altered perception of vertical. Pusher syndrome is sometimes confused with and used interchangeably as the term hemispatial neglect , and some previous theories suggest that neglect leads to pusher syndrome. [2] However, another study had observed that pusher syndrome is also present in patients with left hemisphere lesions, leading to aphasia , providing a stark contrast to what was previously believed regarding hemispatial neglect, which mostly occurs with a right hemisphere lesion. [6] Karnath [2] summarizes these two conflicting views, as they conclude that both neglect and aphasia are highly correlated with pusher syndrome possibly due to the close proximity of relevant brain structures associated with these two respective syndromes. ... Other causes of hemiplegia include spinal cord injury , specifically Brown-Séquard syndrome , traumatic brain injury , or disease affecting the brain . ... "Understanding and treating "pusher syndrome " " . Physical Therapy . 83 (12): 1119–25. doi : 10.1093/ptj/83.12.1119 . ... "Clinical examination tools for lateropulsion or pusher syndrome following stroke: a systematic review of the literature".BCHE, PLAT, ATP1A2, COL4A1, SCN1A, SLC2A1, ALAD, STAT4, KLRC4, TTR, TSC2, TSC1, TPP2, TLR4, TGFBR3, SLC1A3, SMARCB1, CFHR3, RASA1, PRNP, PDGFB, OPA1, NOTCH1, DNM1L, TREX1, PDCD10, DOCK6, TUBB2B, CYP26C1, UBAC2, EOGT, WDR62, IL23R, ANGPTL6, IRF2BPL, ARHGAP31, NF2, TBC1D24, DLL4, ADA2, ERAP1, SUFU, PNPLA8, TBK1, RHOBTB2, NID1, TRNW, FAS, IL12A, TRNV, SP110, HLA-DQB1, HLA-B, CFHR1, CFH, GNAQ, GBE1, GALC, MTOR, FGFR1, ENG, COL3A1, CCR1, CACNA1A, C4A, ATP1A3, IL10, RBPJ, KRAS, ND5, TRNS2, TRNS1, TRNQ, TRNL1, TRNK, MEFV, ND6, TRNF, ND1, CYTB, COX3, COX2, COX1, TRNC, IL12A-AS1, FAP, ENPEP, MMP9, BDNF, NOTCH3, CIMT
-
Nystagmus
Wikipedia
It can be insular or accompany other disorders (such as micro-ophthalmic anomalies or Down syndrome ). Early-onset nystagmus itself is usually mild and non-progressive. ... Types of early-onset nystagmus include the following, along with some of their causes: Infantile: Albinism Aniridia Bilateral congenital cataract Bilateral optic nerve hypoplasia Idiopathic Leber's congenital amaurosis Optic nerve or macular disease Persistent tunica vasculosa lentis Rod monochromatism Visual-motor syndrome of functional monophthalmus Latent nystagmus Noonan syndrome Nystagmus blockage syndrome X-linked infantile nystagmus is associated with mutations of the gene FRMD7 , which is located on the X chromosome . [6] [7] Infantile nystagmus is also associated with two X-linked eye diseases known as complete congenital stationary night blindness (CSNB) and incomplete CSNB (iCSNB or CSNB-2), which are caused by mutations of one of two genes located on the X chromosome. ... "Gaze-evoked and rebound nystagmus in a cerebellar syndrome" . Neuro-Ophthalmology Virtual Education Library (NOVEL): Daniel Gold Collection . ... "Improvement in visual acuity following surgery for correction of head posture in infantile nystagmus syndrome". Journal of Pediatric Ophthalmology and Strabismus . 48 (6): 341–6. doi : 10.3928/01913913-20110118-02 . ... "Effects of tenotomy on patients with infantile nystagmus syndrome: foveation improvement over a broadened visual field".ATXN2, CACNA1A, VLDLR, TDP1, APTX, WWOX, COQ2, ATXN10, PIK3R5, SETX, AFG3L2, STUB1, XPA, TBP, CACNB4, SPTBN2, SLC1A3, ATXN1, RFC1, PRKCG, MRE11, ATXN3, GRID2, FGF14, SPG11, LY6E
-
Chromosome 16p12.1 Deletion Syndrome, 520-Kb
Omim
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome (chr16:21.85-22.37 Mb). Description There are several phenotypes associated with variation in pericentric region of chromosome 16: see the 16p12.2-p11.2 deletion syndrome (613604); see 611913 for a deletion or duplication at 16p11.2 associated with autism (AUTS14); and see 613444 for a 220-kb deletion at 16p11.2 associated with isolated severe early-onset obesity and obesity with developmental delay. ... Seven of 21 had congenital cardiac disease, including 4 with a hypoplastic left heart syndrome. Seizure disorders were observed in 8 of 22 cases, and hypotonia was present in 10 of 21 cases. ... These included the 16p12.1 deletion, the 16p11.2 duplication (614671), and the 15q11.2 deletion (608636). They found that syndromic disorders could be distinguished from those with extreme phenotypic heterogeneity on the basis of the total number of copy number variants and whether the variants are inherited or de novo.
-
Cryptophthalmos, Unilateral Or Bilateral, Isolated
Omim
Mutation in the FREM2 gene can also cause a syndromic form of cryptophthalmos, Fraser syndrome-2 (FRASRS2; 617666). ... Clinical Features Thomas et al. (1986) reviewed syndromic and isolated cryptophthalmos. The principal syndromic form is Fraser syndrome (219000), a recessive. ... The dominant inheritance distinguished the condition from the Fraser syndrome. Saal et al. (1992) concluded that the same condition was present in the patients reported by Coover (1910, 1910, 1915) and Magruder (1921). ... No mutations were detected in other ocular morphogenesis- or Fraser syndrome-associated genes. INHERITANCE - Autosomal recessive HEAD & NECK Face - Continuous intact skin covering upper half of face Eyes - No eyebrows - No eyelashes - No eyelids - No anatomic eyelid structure seen at surgery - Gourd-shaped cyst in ocular cavity - No clear internal structures - Extraocular muscles in narrow orbital fat gap - Optic nerve enlargement in expanded orbit MISCELLANEOUS - Cryptophthalmos may be unilateral or bilateral - Normal brain morphology MOLECULAR BASIS - Caused by mutation in the FRAS1-related extracellular matrix protein-2 gene (FREM2, 608945.0004 ) ▲ Close
-
Pycnodysostosis
Wikipedia
Find sources: "Pycnodysostosis" – news · newspapers · books · scholar · JSTOR ( February 2020 ) ( Learn how and when to remove this template message ) ( Learn how and when to remove this template message ) Pycnodysostosis Pycnodysostosis is inherited in an autosomal recessive manner Specialty Rheumatology , medical genetics , endocrinology Pycnodysostosis (from Greek : πυκνός (puknos) meaning "dense", [1] dys ("defective"), and ostosis ("condition of the bone")), is a lysosomal storage disease of the bone caused by a mutation in the gene that codes the enzyme cathepsin K . [2] Contents 1 Signs and symptoms 2 Genetics 3 Diagnosis 4 Treatment 5 Epidemiology 6 References 7 External links Signs and symptoms [ edit ] Pycnodysostosis causes the bones to be abnormally dense ( osteopetrosis ); the last bones of the fingers (the distal phalanges ) to be unusually short; and delays the normal closure of the connections ( sutures ) of the skull bones in infancy, so that the "soft spot" ( fontanelle ) on top of the head remains widely open. [3] Those with the syndrome have brittle bones which easily break , especially in the legs and feet. ... The height of adult males with the disease is less than 150 cm (59 in). Adult females with the syndrome are even shorter. The disease is also known as Toulouse-Lautrec Syndrome, after the French artist Henri de Toulouse-Lautrec , who may have had the disease. ... External links [ edit ] Classification D ICD - 10 : Q78.8 OMIM : 265800 MeSH : D058631 External resources Orphanet : 763 v t e Bone and joint disease Bone Inflammation endocrine : Osteitis fibrosa cystica Brown tumor infection : Osteomyelitis Sequestrum Involucrum Sesamoiditis Brodie abscess Periostitis Vertebral osteomyelitis Metabolic Bone density Osteoporosis Juvenile Osteopenia Osteomalacia Paget's disease of bone Hypophosphatasia Bone resorption Osteolysis Hajdu–Cheney syndrome Ainhum Gorham's disease Other Ischaemia Avascular necrosis Osteonecrosis of the jaw Complex regional pain syndrome Hypertrophic pulmonary osteoarthropathy Nonossifying fibroma Pseudarthrosis Stress fracture Fibrous dysplasia Monostotic Polyostotic Skeletal fluorosis bone cyst Aneurysmal bone cyst Hyperostosis Infantile cortical hyperostosis Osteosclerosis Melorheostosis Pycnodysostosis Joint Chondritis Relapsing polychondritis Other Tietze's syndrome Combined Osteochondritis Osteochondritis dissecans Child leg: hip Legg–Calvé–Perthes syndrome tibia Osgood–Schlatter disease Blount's disease foot Köhler disease Sever's disease spine Scheuermann's_disease arm: wrist Kienböck's disease elbow Panner disease

-
Acanthocyte
Wikipedia
They are seen on blood films in, among others abetalipoproteinemia , [3] liver disease, chorea acanthocytosis , McLeod syndrome , and several inherited neurological and other disorders, such as neuroacanthocytosis , [4] anorexia nervosa , infantile pyknocytosis , hypothyroidism , idiopathic neonatal hepatitis , alcoholism , congestive splenomegaly , Zieve syndrome , and chronic granulomatous disease . [5] Contents 1 Usage 2 Pathophysiology 3 Differential diagnoses 4 See also 5 References 6 External links Usage [ edit ] Spur cells may refer synonymously to acanthocytes, [6] or may refer in some sources to a specific subset of 'extreme acanthocytes' that have undergone splenic modification whereby additional cell membrane loss has blunted the spicules and the cells have become spherocytic ('spheroacanthocyte'), as seen in some patients with severe liver disease. [7] Acanthocytosis can refer generally to the presence of this type of crenated red blood cell, such as may be found in severe cirrhosis or pancreatitis, [6] but can refer specifically to abetalipoproteinemia , a clinical condition with acanthocytic red blood cells, neurologic problems and steatorrhea. [8] This particular cause of acanthocytosis (also known as abetalipoproteinemia, apolipoprotein B deficiency, and Bassen-Kornzweig syndrome) is a rare, genetically inherited, autosomal recessive condition due to the inability to fully digest dietary fats in the intestines as a result of various mutations of the microsomal triglyceride transfer protein (MTTP) gene. [9] Pathophysiology [ edit ] Acanthocytosis in a patient with abetalipoproteinemia Acanthocytes arise from either alterations in membrane lipids or structural proteins. ... Alteration in membrane structural proteins are seen in neuroacanthocytosis and McLeod syndrome. In liver dysfunction, apolipoprotein A-II deficient lipoprotein accumulates in plasma causing increased cholesterol in RBCs. ... In abetalipoproteinemia, there is deficiency of lipids and vitamin E causing abnormal morphology of RBCs. [10] Differential diagnoses [ edit ] The diagnosis of Acanthocytosis should be differentiated from: acute or chronic anemia, hepatitis A, B, and C, hepatorenal syndrome , hypopituitarism, malabsorption syndromes, and malnutrition [11] Acanthocytosis secondary to malnourishment, such as anorexia nervosa and cystic fibrosis, remits with resolution of the nutritional deficiency. [12] Acanthocyte-like cells may be found in hypothyroidism, after splenectomy, and in myelodysplasia. [12] Acanthocytes should be distinguished from echinocytes , which are also called 'burr cells', which although crenated are dissimilar in that they have multiple, small, projecting spiculations at regular intervals on the cell membrane. [7] [12] Burr cells usually imply uremia , but are seen in many conditions, including mild hemolysis in hypomagnesemia and hypophosphatemia, hemolytic anemia in long-distance runners, and pyruvate kinase deficiency . [11] Burr cells can also arise in vitro due to elevated pH, blood storage, ATP depletion, calcium accumulation, and contact with glass. [11] Acanthocytes should also be distinguished from keratocytes, also called 'horn cells' which have a few very large protuberances. [12] See also [ edit ] List of hematologic conditions References [ edit ] ^ " acanthocyte " at Dorland's Medical Dictionary ^ Wrongdiagnosis --> Acanthocytosis Retrieved on October 12, 2009 ^ Cooper RA, Durocher JR, Leslie MH (July 1977). ... (August 4, 2011). "Bassen-Kornzweig syndrome" . Pub Med Health . National Center for Biotechnology Information, U.S.

-
Musical Ear Syndrome
Wikipedia
Auditory hallucination associated with hearing loss Musical ear syndrome ( MES ) describes a condition seen in people who have hearing loss and subsequently develop auditory hallucinations . "MES" has also been associated with musical hallucinations , which is a complex form of auditory hallucinations where an individual may experience music or sounds that are heard without an external source. [1] It is comparable to Charles Bonnet syndrome (visual hallucinations in visually impaired people) [2] and some have suggested this phenomenon could be included under this diagnosis. [3] Contents 1 Causes 2 Treatment 3 Populations 4 History 5 References Causes [ edit ] It is postulated that by the " release phenomenon " MES is caused by hypersensitivity in the auditory cortex caused by sensory deprivation, secondary to their hearing loss. [4] This "hole" in the hearing range is "plugged" by the brain confabulating a piece of information – in this case a piece of music. ... PMID 22643207 . ^ Berrios GE, Brook P (1982). "The Charles Bonnet Syndrome and the Problems of Visual Perceptual Disorder in the Elderly". ... "Spontaneous musical auditory perceptions in patients who develop abrupt bilateral sensorineural hearing loss. An uninhibition syndrome?". Acta Oto-Laryngologica . 126 (4): 368–74. doi : 10.1080/00016480500416942 .
-
Salivary Gland Hypoplasia
Wikipedia
Salivary gland hypoplasia is relative underdevelopment of the Salivary glands . [1] Salivary gland hypoplasia tends to produce xerostomia (dry mouth), with all the associated problems this brings. [1] [2] It is a rare condition, [2] which may occur as a congenital abnormality or result from lack of neuromuscular stimulation. [1] It may be associated with Melkersson–Rosenthal syndrome , [1] [2] and hereditary ectodermal dysplasia . [1] References [ edit ] ^ a b c d e Purkait SK (1 June 2011). ... ISBN 978-93-5090-850-1 . v t e Oral and maxillofacial pathology Lips Cheilitis Actinic Angular Plasma cell Cleft lip Congenital lip pit Eclabium Herpes labialis Macrocheilia Microcheilia Nasolabial cyst Sun poisoning Trumpeter's wart Tongue Ankyloglossia Black hairy tongue Caviar tongue Crenated tongue Cunnilingus tongue Fissured tongue Foliate papillitis Glossitis Geographic tongue Median rhomboid glossitis Transient lingual papillitis Glossoptosis Hypoglossia Lingual thyroid Macroglossia Microglossia Rhabdomyoma Palate Bednar's aphthae Cleft palate High-arched palate Palatal cysts of the newborn Inflammatory papillary hyperplasia Stomatitis nicotina Torus palatinus Oral mucosa – Lining of mouth Amalgam tattoo Angina bullosa haemorrhagica Behçet's disease Bohn's nodules Burning mouth syndrome Candidiasis Condyloma acuminatum Darier's disease Epulis fissuratum Erythema multiforme Erythroplakia Fibroma Giant-cell Focal epithelial hyperplasia Fordyce spots Hairy leukoplakia Hand, foot and mouth disease Hereditary benign intraepithelial dyskeratosis Herpangina Herpes zoster Intraoral dental sinus Leukoedema Leukoplakia Lichen planus Linea alba Lupus erythematosus Melanocytic nevus Melanocytic oral lesion Molluscum contagiosum Morsicatio buccarum Oral cancer Benign: Squamous cell papilloma Keratoacanthoma Malignant: Adenosquamous carcinoma Basaloid squamous carcinoma Mucosal melanoma Spindle cell carcinoma Squamous cell carcinoma Verrucous carcinoma Oral florid papillomatosis Oral melanosis Smoker's melanosis Pemphigoid Benign mucous membrane Pemphigus Plasmoacanthoma Stomatitis Aphthous Denture-related Herpetic Smokeless tobacco keratosis Submucous fibrosis Ulceration Riga–Fede disease Verruca vulgaris Verruciform xanthoma White sponge nevus Teeth ( pulp , dentin , enamel ) Amelogenesis imperfecta Ankylosis Anodontia Caries Early childhood caries Concrescence Failure of eruption of teeth Dens evaginatus Talon cusp Dentin dysplasia Dentin hypersensitivity Dentinogenesis imperfecta Dilaceration Discoloration Ectopic enamel Enamel hypocalcification Enamel hypoplasia Turner's hypoplasia Enamel pearl Fluorosis Fusion Gemination Hyperdontia Hypodontia Maxillary lateral incisor agenesis Impaction Wisdom tooth impaction Macrodontia Meth mouth Microdontia Odontogenic tumors Keratocystic odontogenic tumour Odontoma Dens in dente Open contact Premature eruption Neonatal teeth Pulp calcification Pulp stone Pulp canal obliteration Pulp necrosis Pulp polyp Pulpitis Regional odontodysplasia Resorption Shovel-shaped incisors Supernumerary root Taurodontism Trauma Avulsion Cracked tooth syndrome Vertical root fracture Occlusal Tooth loss Edentulism Tooth wear Abrasion Abfraction Acid erosion Attrition Periodontium ( gingiva , periodontal ligament , cementum , alveolus ) – Gums and tooth-supporting structures Cementicle Cementoblastoma Gigantiform Cementoma Eruption cyst Epulis Pyogenic granuloma Congenital epulis Gingival enlargement Gingival cyst of the adult Gingival cyst of the newborn Gingivitis Desquamative Granulomatous Plasma cell Hereditary gingival fibromatosis Hypercementosis Hypocementosis Linear gingival erythema Necrotizing periodontal diseases Acute necrotizing ulcerative gingivitis Pericoronitis Peri-implantitis Periodontal abscess Periodontal trauma Periodontitis Aggressive As a manifestation of systemic disease Chronic Perio-endo lesion Teething Periapical, mandibular and maxillary hard tissues – Bones of jaws Agnathia Alveolar osteitis Buccal exostosis Cherubism Idiopathic osteosclerosis Mandibular fracture Microgenia Micrognathia Intraosseous cysts Odontogenic : periapical Dentigerous Buccal bifurcation Lateral periodontal Globulomaxillary Calcifying odontogenic Glandular odontogenic Non-odontogenic: Nasopalatine duct Median mandibular Median palatal Traumatic bone Osteoma Osteomyelitis Osteonecrosis Bisphosphonate-associated Neuralgia-inducing cavitational osteonecrosis Osteoradionecrosis Osteoporotic bone marrow defect Paget's disease of bone Periapical abscess Phoenix abscess Periapical periodontitis Stafne defect Torus mandibularis Temporomandibular joints , muscles of mastication and malocclusions – Jaw joints, chewing muscles and bite abnormalities Bruxism Condylar resorption Mandibular dislocation Malocclusion Crossbite Open bite Overbite Overeruption Overjet Prognathia Retrognathia Scissor bite Maxillary hypoplasia Temporomandibular joint dysfunction Salivary glands Benign lymphoepithelial lesion Ectopic salivary gland tissue Frey's syndrome HIV salivary gland disease Necrotizing sialometaplasia Mucocele Ranula Pneumoparotitis Salivary duct stricture Salivary gland aplasia Salivary gland atresia Salivary gland diverticulum Salivary gland fistula Salivary gland hyperplasia Salivary gland hypoplasia Salivary gland neoplasms Benign: Basal cell adenoma Canalicular adenoma Ductal papilloma Monomorphic adenoma Myoepithelioma Oncocytoma Papillary cystadenoma lymphomatosum Pleomorphic adenoma Sebaceous adenoma Malignant: Acinic cell carcinoma Adenocarcinoma Adenoid cystic carcinoma Carcinoma ex pleomorphic adenoma Lymphoma Mucoepidermoid carcinoma Sclerosing polycystic adenosis Sialadenitis Parotitis Chronic sclerosing sialadenitis Sialectasis Sialocele Sialodochitis Sialosis Sialolithiasis Sjögren's syndrome Orofacial soft tissues – Soft tissues around the mouth Actinomycosis Angioedema Basal cell carcinoma Cutaneous sinus of dental origin Cystic hygroma Gnathophyma Ludwig's angina Macrostomia Melkersson–Rosenthal syndrome Microstomia Noma Oral Crohn's disease Orofacial granulomatosis Perioral dermatitis Pyostomatitis vegetans Other Eagle syndrome Hemifacial hypertrophy Facial hemiatrophy Oral manifestations of systemic disease This article about a disease , disorder, or medical condition is a stub .
-
Purpura
Wikipedia
External links [ edit ] Classification D ICD - 10 : D69 ICD - 9-CM : 287 MeSH : D011693 DiseasesDB : 25619 External resources MedlinePlus : 003232 Evaluating the Child with Purpura from American Academy of Family Physicians v t e Disorders of bleeding and clotting Coagulation · coagulopathy · Bleeding diathesis Clotting By cause Clotting factors Antithrombin III deficiency Protein C deficiency Activated protein C resistance Protein S deficiency Factor V Leiden Prothrombin G20210A Platelets Sticky platelet syndrome Thrombocytosis Essential thrombocythemia DIC Purpura fulminans Antiphospholipid syndrome Clots Thrombophilia Thrombus Thrombosis Virchow's triad Trousseau sign of malignancy By site Deep vein thrombosis Bancroft's sign Homans sign Lisker's sign Louvel's sign Lowenberg's sign Peabody's sign Pratt's sign Rose's sign Pulmonary embolism Renal vein thrombosis Bleeding By cause Thrombocytopenia Thrombocytopenic purpura : ITP Evans syndrome TM TTP Upshaw–Schulman syndrome Heparin-induced thrombocytopenia May–Hegglin anomaly Platelet function adhesion Bernard–Soulier syndrome aggregation Glanzmann's thrombasthenia platelet storage pool deficiency Hermansky–Pudlak syndrome Gray platelet syndrome Clotting factor Hemophilia A/VIII B/IX C/XI von Willebrand disease Hypoprothrombinemia/II Factor VII deficiency Factor X deficiency Factor XII deficiency Factor XIII deficiency Dysfibrinogenemia Congenital afibrinogenemia Signs and symptoms Bleeding Bruise Hematoma Petechia Purpura Nonthrombocytopenic purpura By site head Epistaxis Hemoptysis Intracranial hemorrhage Hyphema Subconjunctival hemorrhage torso Hemothorax Hemopericardium Pulmonary hematoma abdomen Gastrointestinal bleeding Hemobilia Hemoperitoneum Hematocele Hematosalpinx joint HemarthrosisPTPN22, CTLA4, ACP5, HIRA, MVK, MYD88, NFKB1, NFKB2, PRKCD, PROC, PROS1, PRTN3, RREB1, STIM1, TBX1, TFR2, TNFSF12, UFD1, JAK2, SEC24C, SH2B3, CFHR3, TNFRSF13B, ICOS, ADA2, NLRC4, ORAI1, NLRP3, TNFRSF13C, ARL6IP6, MPL, JMJD1C, ITGB3, MS4A1, GP1BA, GP1BB, GP9, CFH, CFHR1, COMT, CALR, HLA-DPA1, HLA-DPB1, CD81, CR2, C4A, ITGA2B, C2, ARVCF, CD19, ADAMTS13, HLA-DRB1, SMUG1, C4B, CAT, EDN1, CP, CTNNB1, NPHS2, MEFV, GLUL, HLA-A, HLA-B, IL1B, IL6, MMP9, AGT, MET









