-
Deafness, Autosomal Recessive 18a
Omim
Mutations in the harmonin gene also cause Usher syndrome type IC (USH1C; 276904). A form of autosomal recessive deafness designated DFNB18B (614945), which also maps to chromosome 11p15, is caused by mutation in the OTOG gene (604487). ... Based on the linkage data, Jain et al. (1997, 1998) postulated that DFNB18 and USH1C are allelic. (Usher syndrome, of which there are many forms, has retinopathy in association with deafness.) ... Mutations in connexin-26 (121011) provide another example of syndrome deafness and nonsyndromic deafness being caused by different alleles of the same gene: isolated autosomal recessive deafness (220290), autosomal dominant isolated deafness (601544), and keratoderma with deafness (148350). In the family in which Jain et al. (1998) mapped DFNB18 to 11p, Ahmed et al. (2002) identified a leaky splice site mutation in the harmonin gene (605242.0008), indicating that Usher syndrome type IC and DFNB18 are allelic. ... Harmonin has been shown to bind, by means of its PDZ-domains, with the products of other Usher syndrome genes, including Myo7a, Cdh23 (605516), and Sans (USH1G; 607696).
-
Short-Rib Thoracic Dysplasia 17 With Or Without Polydactyly
Omim
SRTD encompasses Ellis-van Creveld syndrome (EVC) and the disorders previously designated as Jeune syndrome or asphyxiating thoracic dystrophy (ATD), short rib-polydactyly syndrome (SRPS), and Mainzer-Saldino syndrome (MZSDS). ... There is phenotypic overlap with the cranioectodermal dysplasias (Sensenbrenner syndrome; see CED1, 218330). For a discussion of genetic heterogeneity of short-rib thoracic dysplasia with or without polydactyly, see SRTD1 (208500).
-
Congenital Membranous Nephropathy Due To Fetomaternal Anti-Neutral Endopeptidase Alloimmunization
Orphanet
A rare, congenital glomerular disease due to maternal anti-neutral endopeptidase (NEP) alloimmunization characterized by severe renal failure and nephrotic syndrome at birth, which rapidly improves in the first weeks of life. ... Clinical description Presentation is at birth with nephrotic syndrome, acute renal failure (oligoanuria), or both. ... Electron microscopy examination shows abundant electron-dense deposits typically containing annular formations on the outer aspect of the glomerular capillary wall and a marked atrophy of the brush border. Differential diagnosis The syndrome should be differentiated from other causes of early-onset membranous nephropathy such as congenital infections like syphilis and toxoplasmosis, and neonatal lupus erythematosus. ... In neonates with severe presentation, i.e. renal failure and/or nephrotic syndrome, an exchange transfusion to eliminate circulating anti-NEP antibodies can be considered. ... Prognosis Infants usually show a rapid improvement of renal failure and the nephrotic syndrome, although a severe form requiring prolonged dialysis may also be observed.
-
Lung Disease, Immunodeficiency, And Chromosome Breakage Syndrome
Omim
A number sign (#) is used with this entry because of evidence that lung disease, immunodeficiency, and chromosome breakage syndrome (LICS) is caused by homozygous or compound heterozygous mutation in the NSMCE3 gene (608243) on chromosome 15q13. Description LICS is an autosomal recessive chromosome breakage syndrome characterized by failure to thrive in infancy, immune deficiency, and fatal progressive pediatric lung disease induced by viral infection.
-
Mitochondrial Dna Depletion Syndrome 12a (Cardiomyopathic Type), Autosomal Dominant
Omim
A number sign (#) is used with this entry because of evidence that autosomal dominant mitochondrial DNA depletion syndrome-12A (MTDPS12A) is caused by heterozygous mutation in the SLC25A4 gene (103220) on chromosome 4q35. ... For a discussion of genetic heterogeneity of mtDNA depletion syndromes, see MTDPS1 (603041). Clinical Features Thompson et al. (2016) reported 7 children from 6 unrelated families who presented at birth with profound hypotonia, little spontaneous movement, lactic acidosis, and respiratory insufficiency necessitating mechanical ventilation.
-
Al Kaissi Syndrome
Omim
A number sign (#) is used with this entry because of evidence that Al Kaissi syndrome (ALKAS) is caused by homozygous mutation in the CDK10 gene (603464) on chromosome 16q24. Description Al Kaissi syndrome is an autosomal recessive developmental disorder characterized by growth retardation, spine malformation, particularly of the cervical spine, dysmorphic facial features, and delayed psychomotor development with moderate to severe intellectual disability (summary by Windpassinger et al., 2017).
-
Macrodystrophia Lipomatosa
Wikipedia
Fibrolipomatous hamartoma Proteus syndrome [3] Neurofibromatosis type 1. [4] Klippel Trenaunay syndrome . [5] Parkes Weber syndrome Hemangiomas . [6] Management [ edit ] No medical therapy exists for such a disorder. ... "Orthopaedic manifestations of Proteus syndrome in a child with literature update" . ... "Clinical experience of the Klippel-Trenaunay syndrome" . Arch Plast Surg . 42 (5): 552–8. doi : 10.5999/aps.2015.42.5.552 .
-
Paradoxical Embolism
Wikipedia
PMID 17433144 . [ dead link ] External links [ edit ] Classification D MeSH : D019320 SNOMED CT : 134296002 External resources eMedicine : article/460607 v t e Cardiovascular disease (vessels) Arteries , arterioles and capillaries Inflammation Arteritis Aortitis Buerger's disease Peripheral artery disease Arteriosclerosis Atherosclerosis Foam cell Fatty streak Atheroma Intermittent claudication Critical limb ischemia Monckeberg's arteriosclerosis Arteriolosclerosis Hyaline Hyperplastic Cholesterol LDL Oxycholesterol Trans fat Stenosis Carotid artery stenosis Renal artery stenosis Other Aortoiliac occlusive disease Degos disease Erythromelalgia Fibromuscular dysplasia Raynaud's phenomenon Aneurysm / dissection / pseudoaneurysm torso : Aortic aneurysm Abdominal aortic aneurysm Thoracic aortic aneurysm Aneurysm of sinus of Valsalva Aortic dissection Aortic rupture Coronary artery aneurysm head / neck Intracranial aneurysm Intracranial berry aneurysm Carotid artery dissection Vertebral artery dissection Familial aortic dissection Vascular malformation Arteriovenous fistula Arteriovenous malformation Telangiectasia Hereditary hemorrhagic telangiectasia Vascular nevus Cherry hemangioma Halo nevus Spider angioma Veins Inflammation Phlebitis Venous thrombosis / Thrombophlebitis primarily lower limb Deep vein thrombosis abdomen Hepatic veno-occlusive disease Budd–Chiari syndrome May–Thurner syndrome Portal vein thrombosis Renal vein thrombosis upper limb / torso Mondor's disease Paget–Schroetter disease head Cerebral venous sinus thrombosis Post-thrombotic syndrome Varicose veins Gastric varices Portacaval anastomosis Caput medusae Esophageal varices Hemorrhoid Varicocele Other Chronic venous insufficiency Chronic cerebrospinal venous insufficiency Superior vena cava syndrome Inferior vena cava syndrome Venous ulcer Arteries or veins Angiopathy Macroangiopathy Microangiopathy Embolism Pulmonary embolism Cholesterol embolism Paradoxical embolism Thrombosis Vasculitis Blood pressure Hypertension Hypertensive heart disease Hypertensive emergency Hypertensive nephropathy Essential hypertension Secondary hypertension Renovascular hypertension Benign hypertension Pulmonary hypertension Systolic hypertension White coat hypertension Hypotension Orthostatic hypotension This article about a medical condition affecting the circulatory system is a stub .
-
Keratoconus
Medlineplus
Keratoconus can be a feature of genetic syndromes, such as Leber congenital amaurosis and arterial tortuosity syndrome. When it is part of a syndrome, keratoconus is caused by the same genetic mutation that causes the syndrome. Mutations in the genes that cause syndromes with keratoconus have not been found to cause keratoconus without other features.
-
Lymphangiectasia, Pulmonary, Congenital
Omim
Jacquemont et al. (2000) noted the resemblance to Hennekam syndrome (235510), but concluded that their cases and the cases reported by Njolstad et al. (1998) represented a distinct nosologic entity. ... CPL may also be part of a multiple congenital anomalies (MCA) syndrome, such as Noonan syndrome (163950), Turner syndrome, and Down syndrome.
-
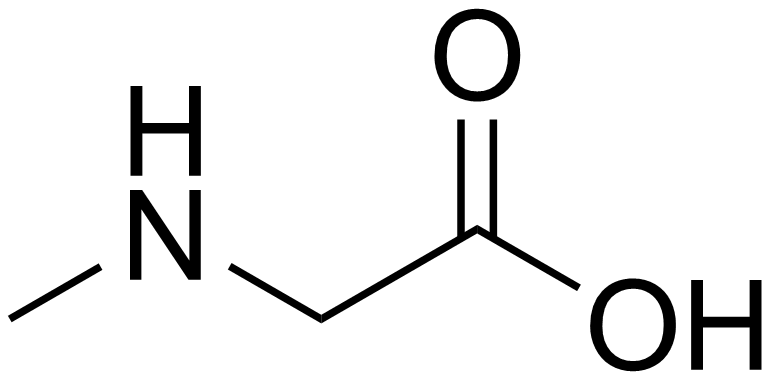
Sarcosinemia
Wikipedia
External links [ edit ] Sarcosinemia at NIH 's Office of Rare Diseases Classification D ICD - 10 : E72.5 ICD - 9-CM : 270.8 OMIM : 268900 MeSH : C537236 DiseasesDB : 29841 External resources Orphanet : 3129 v t e Inborn error of amino acid metabolism K → acetyl-CoA Lysine /straight chain Glutaric acidemia type 1 type 2 Hyperlysinemia Pipecolic acidemia Saccharopinuria Leucine 3-hydroxy-3-methylglutaryl-CoA lyase deficiency 3-Methylcrotonyl-CoA carboxylase deficiency 3-Methylglutaconic aciduria 1 Isovaleric acidemia Maple syrup urine disease Tryptophan Hypertryptophanemia G G→ pyruvate → citrate Glycine D-Glyceric acidemia Glutathione synthetase deficiency Sarcosinemia Glycine → Creatine : GAMT deficiency Glycine encephalopathy G→ glutamate → α-ketoglutarate Histidine Carnosinemia Histidinemia Urocanic aciduria Proline Hyperprolinemia Prolidase deficiency Glutamate / glutamine SSADHD G→ propionyl-CoA → succinyl-CoA Valine Hypervalinemia Isobutyryl-CoA dehydrogenase deficiency Maple syrup urine disease Isoleucine 2-Methylbutyryl-CoA dehydrogenase deficiency Beta-ketothiolase deficiency Maple syrup urine disease Methionine Cystathioninuria Homocystinuria Hypermethioninemia General BC / OA Methylmalonic acidemia Methylmalonyl-CoA mutase deficiency Propionic acidemia G→ fumarate Phenylalanine / tyrosine Phenylketonuria 6-Pyruvoyltetrahydropterin synthase deficiency Tetrahydrobiopterin deficiency Tyrosinemia Alkaptonuria / Ochronosis Tyrosinemia type I Tyrosinemia type II Tyrosinemia type III / Hawkinsinuria Tyrosine → Melanin Albinism : Ocular albinism ( 1 ) Oculocutaneous albinism ( Hermansky–Pudlak syndrome ) Waardenburg syndrome Tyrosine → Norepinephrine Dopamine beta hydroxylase deficiency reverse: Brunner syndrome G→ oxaloacetate Urea cycle / Hyperammonemia ( arginine aspartate ) Argininemia Argininosuccinic aciduria Carbamoyl phosphate synthetase I deficiency Citrullinemia N-Acetylglutamate synthase deficiency Ornithine transcarbamylase deficiency / translocase deficiency Transport / IE of RTT Solute carrier family : Cystinuria Hartnup disease Iminoglycinuria Lysinuric protein intolerance Fanconi syndrome : Oculocerebrorenal syndrome Cystinosis Other 2-Hydroxyglutaric aciduria Aminoacylase 1 deficiency Ethylmalonic encephalopathy Fumarase deficiency Trimethylaminuria
-
Sarcoidosis
Orphanet
Clinical presentation is typically with persistent dry cough, eye or skin manifestations, peripheral lymph nodes, fatigue, weight loss, fever or night sweats, and Löfgren syndrome. Epidemiology Sarcoidosis is a ubiquitous disease with incidence varying according to age, sex, race and geographic origin. ... In most cases, sarcoidosis is revealed by persistent dry cough, eye or skin manifestations, peripheral lymph nodes, fatigue, weight loss, fever or night sweats, and Löfgren syndrome (an acute form characterized by fever, bilateral ankle arthritis and/or erythema nodosum and bilateral hilar lymphadenopathy). ... African-Americans present a more severe disease, and in Scandinavian countries, about one third of cases have Löfgren syndrome. Etiology The etiology remains unknown. ... Some situations do not require biopsy such as Lofgren's syndrome or the presence of typical asymptomatic bilateral mediastino-hilar adenopathies. ... Discomfort may be due to fibrosis, para-sarcoidosis syndrome or comorbidities as well as active disease.HLA-DRB1, BTNL2, ANXA11, IL18, HLA-DPB1, TGFB1, SLC11A1, SOD2, HLA-DQB1, HLA-DQA1, HLA-DRA, HLA-B, HLA-DMB, TYK2, HLA-C, ACE, APOM, HLA-V, ATF6B, ZSCAN12P1, CYP21A2, HLA-DOB, RBM45, STK19, FAM117B, C1orf141, GTF2H4, NOTCH4, IFNG, SMUG1, ZSCAN12, NOD2, MANBA, SP140, SLC27A5, SLC44A4, PARP9, TSBP1, ATXN2, OR5V1, ZSCAN16-AS1, C6orf15, USP8P1, HLA-DRB9, ATP8A2, VARS1, HLA-DRB3, TNF, LINC01149, TSBP1-AS1, LINC02571, CDH4, HLA-DQA2, CEL, OR12D3, MTCO3P1, LTA, IL2, IL1B, CHIT1, VEGFA, IL6, CCL5, TLR4, CCR2, CAT, CD14, PPARG, IL10, PTGS2, TLR2, CHI3L1, IL1A, CCR5, HLA-A, LYZ, IGF1, CR1, COPD, TRBV20OR9-2, SPP1, FCGR3B, FCGR3A, VDR, CFTR, CCL2, PDCD1, CSF2, SOAT1, CYP27B1, CCL3, IL17A, HSPA1L, MIF, VIM, FCGR2A, IL33, TAP2, FOXP3, FLT1, CXCL8, CXCL9, IL5, IL4R, SCGB1A1, IL4, IL2RA, STAT1, IL1RN, CXCL5, IL13, CCL17, CCL8, USO1, GPR162, PSMB8, PON1, FCGR2C, CXCL10, CD163, NCAM1, SEC14L2, MRC1, NXF1, MMP12, TGFB3, TGFB2, IFNA17, HLA-DRB4, AGER, AGT, AGTR1, ALB, FAS, WG, MIR34A, CD40, CDKN1A, HT, FLNB, FOLH1, HIF1A, ESAT, HSPA1B, HSPA1A, HLA-G, NUP214, MIR33A, MIR200B, MIR126, RAB23, WNT3, IL31, YWHAZ, CXCR4, ST8SIA4, ARHGEF5, SGSM3, ACKR4, NCOA3, RECK, PIK3R3, CCL15-CCL14, H3P44, SOCS1, DNAH11, TNFRSF25, IL4I1, TNFSF13, ADAM9, TCL1A, MUC5B, C10orf67, ERVK-12, MTCO2P12, TIMP2, IL19, ING4, TLR1, NT5C3A, TLR3, TLR7, OPN1MW3, ERVK-11, ERVK-22, DDX41, OPN1MW2, TNFRSF1A, TNFRSF1B, TNNI3, ERVK-2, HSP90B2P, TRH, RPL17-C18orf32, TSC2, TWIST1, H3C9P, CD24, RIPK2, OTUD1, KRT20, CXCL16, TLR10, VTCN1, POLG2, MARCHF7, CUL9, IL25, ICOSLG, SEMA4A, IGAN1, TMEM131, ACE2, IL17RA, PPARGC1A, LMOD1, NOCT, PTPN23, PHGDH, GREM1, HAVCR1, RTN4, KNL1, MYDGF, XAF1, ADAMDEC1, CD274, TNFSF13B, IL18R1, TCL1B, DPM2, PLB1, TIMELESS, KALRN, IL23R, TLR9, MBL3P, ZFYVE9, BAG3, ABCG1, CD200R1, SPECC1, ICOS, INTS4, SPATA2, SEC16A, TLR6, TSLP, NOD1, NDC80, HAVCR2, OBSCN, ANP32B, POSTN, ACTB, PSMD12, TERF1, GC, NR3C1, CXCR3, GNAO1, GLRX, GLI1, GLB1, OPN1MW, FOS, GSTP1, FLG, FGFR1, FGF13, FGF2, FCGR2B, FCGR1A, FABP4, GSTM1, GSTT1, IL7, HP, IGHG1, IFNA13, IFNA10, IFNA2, IFNA1, IFN1@, TNC, HNRNPD, GZMB, HMGB1, HLA-DQB2, HLA-DPA1, HLA-DOA, HLA-DMA, HGF, HFE, F13A1, F2R, ERG, AQP4, CD3G, BRAF, BMPR2, BDNF, BCL2, BBS2, B2M, APOA1, EPHX2, XIAP, APEH, ANXA2, ANXA1, ALK, AGTR2, ADA, MS4A1, CD80, CD86, TNFRSF8, EPHA2, EGR1, EDN1, CYBB, CX3CR1, CTNNB1, CSF1, CRP, COL3A1, COL1A2, COL1A1, CCR7, CMA1, CD69, CD68, IGKC, IL7R, TAP1, PTHLH, SAA1, S100A9, S100A8, RTN1, RREB1, RPL32, RPL17, PTH, SAT1, ACTN4, PSMB9, PSMB2, PRL, MAP2K7, PLA2G1B, PIK3CG, SAA2, CCL1, IL9, SELE, TACR1, TAC1, STAT4, STAT3, SNRNP70, FSCN1, SNCA, CX3CL1, CCL13, CXCL11, CCL22, CCL20, CCL19, CCL18, CCL16, CCL15, PIK3CD, PIK3CB, PIK3CA, KDR, MCAM, MBL2, SMAD3, LPA, LGALS1, LCN2, KIR2DL1, ITGAX, PDGFA, ITGAM, ITGAE, IRF5, IRF4, IL15, IL11, IL9R, MEFV, CIITA, MIP, MMP1, PAM, P4HB, ORM1, TNFRSF11B, NCL, MYD88, MUC1, COX2, MST1, MPP1, MNAT1, MMP14, MMP9, MMP7, MMP2, LINC02605

-
Mosaic Genome-Wide Paternal Uniparental Disomy
Orphanet
A rare chromosomal anomaly characterized by a combination of paternal uniparental and biparental cell lineages, leading to variable clinical presentation that predominantly includes features of Beckwith-Wiedemann syndrome and increased risk of various tumors. In addition, features of Angelman syndrome and transient neonatal diabetes might be expected.
-
Mycobacterium Avium-Intracellulare Infection
Wikipedia
Since the six patients in their retrospective case series were older females, Reich and Johnson proposed that patients without a vigorous cough may develop right middle lobe or left lingular infection with MAC. They proposed this syndrome be named Lady Windermere syndrome, after the character Lady Windermere in Oscar Wilde 's play Lady Windermere's Fan . ... There is no evidence that a person's reluctance to spit has any causal role in NTM infection, the chief reason for the term having been applied to older women presenting with the condition. [22] Lady Windermere syndrome is a type of mycobacterial lung infection. [23] Literary reference [ edit ] The original Chest article proposing the existence and pathophysiology of the Lady Windermere syndrome suggested the character Lady Windermere in Oscar Wilde 's Victorian-era play Lady Windermere's Fan is a good example of the fastidious behavior believed to cause the syndrome. ... Shortly after the Lady Windermere syndrome was proposed, a librarian wrote a letter to the editor of Chest [24] challenging the use of Lady Windermere as the eponymous ancestor of the proposed syndrome. ... (August 2018). "In Defense of Lady Windermere Syndrome" . Lung . 196 (4): 377–379. doi : 10.1007/s00408-018-0122-x . ... PMID 18515557 . ^ Kasthoori JJ, Liam CK, Wastie ML (February 2008). "Lady Windermere syndrome: an inappropriate eponym for an increasingly important condition".HSPD1, TNF, SLC11A1, IFNG, NTM, STAT1, STAT4, TLR2, TNFSF4, TP63, TNFSF9, ARHGEF2, IL32, AIM2, RABEPK, LANCL1, STS, PSMD7, IL37, PYCARD, IL23A, NCAPG2, RGMA, MLIP, NLRP3, IL31RA, ARMH1, MIR346, AK6, EBNA1BP2, MAPK1, CAMP, IFNA13, CASP1, TNFSF8, CD70, CFTR, CCR5, FCN2, GATA2, HLA-B, HSPE1, ICAM1, IFNA1, IFNB1, NMT1, IL1B, IL2, IL9, IL12RB1, TNFRSF9, IL17A, CXCL10, RPSA, CXCL9, MT1JP, RNR1, H3P28

-
Diverticulosis
Wikipedia
However, it is unclear if these symptoms are attributable to the underlying diverticulosis or to coexistent irritable bowel syndrome . [7] Diverticular disease was found associated with a higher risk of left sided colon cancer. [8] Bleeding [ edit ] Diverticular disease can present with painless rectal bleeding as bright red blood per rectum. ... National Institutes of Health notes that, although the low-fiber theory of the cause of diverticulosis is the leading theory, it has not yet been proven. [17] Genetics [ edit ] The predisposition to diverticulosis for specific individuals is likely explained by a genetic component, a theory which is supported by studies examining the rates of diverticulosis among twins. [1] The heritability of diverticulosis is estimated to be approximately 40%. [1] Intestinal motility [ edit ] Another theory suggests the degeneration of glial neurons in the myenteric plexus and the interstitial cells of Cajal lead to slowed intestinal movement and consequently fecal content exerts increased pressure on the colon wall resulting in the formation of diverticula. [1] Risk factors [ edit ] Advanced age constipation a diet that is low in dietary fiber (although this claim is controversial) connective tissue disorders (such as Marfan syndrome and Ehlers Danlos Syndrome ) that may cause weakness in the colon wall hereditary or genetic predisposition , [18] extreme weight loss heavy meat consumption [19] [20] [21] Pathophysiology [ edit ] Drawing showing a sigmoid colon with many diverticula The precise mechanisms by which diverticula are formed are unknown. [1] Multiple theories have been proposed including genetic susceptibility, diet, intestinal motility, changes in the microbiome , and inflammation . ... Diverticular disease is more common in collagen disorders such as Ehlers Danlos Syndrome. [29] History [ edit ] The modern emphasis on the value of fiber in the diet began with Thomas L. ... Diverticulosis —PrimeHealthChannel v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum

-
Tumors Of The Hematopoietic And Lymphoid Tissues
Wikipedia
Lymphomas, lymphocytic leukemias, and myeloma are from the lymphoid line, while acute and chronic myelogenous leukemia, myelodysplastic syndromes and myeloproliferative diseases are myeloid in origin. ... However, the influential WHO Classification (published in 2001 and updated in 2008 and 2016) places a greater emphasis on cell lineage. [ citation needed ] Relative proportions of hematological malignancies in the United States [5] Type of hematological malignancy Percentage Total Leukemias — 30.4% Acute lymphoblastic leukemia (ALL) 4.0% Acute myelogenous leukemia (AML) 8.7% Chronic lymphocytic leukemia (CLL) sorted under lymphomas according to current WHO classification; called small lymphocytic lymphoma (SLL) when leukemic cells are absent. 10.2% Chronic myelogenous leukemia (CML) 3.7% Acute monocytic leukemia (AMoL) 0.7% Other leukemias 3.1% Lymphomas — 55.6% Hodgkin's lymphomas (all four subtypes) 7.0% Non-Hodgkin's lymphomas (all subtypes) 48.6% Myelomas 14.0% Total 100% Classification according to WHO [ edit ] 4th Edition [6] NOS = "Not otherwise specified" Myeloid neoplasms Myeloproliferative neoplasms Chronic myeloid leukaemia, BCR-ABL1-positive Chronic neutrophilic leukaemia Polycythaemia vera Primary myelofibrosis Essential thrombocythaemia Chronic eosinophilic leukaemia, NOS Myeloproliferative neoplasm, unclassifiable Mastocytosis Cutaneous mastocytosis Indolent systemic mastocytosis Systemic mastocytosis with an associated haematological neoplasm Aggressive systemic mastocytosis Mast cell leukaemia Mast cell sarcoma Myeloid/lymphoid neoplasms with eosinophilia and gene rearrangement Myeloid/lymphoid neoplasms with PDGFRA rearrangement Myeloid/lymphoid neoplasms with PDGFRB rearrangement Myeloid/lymphoid neoplasms with FGFR1 rearrangement Myeloid/lymphoid neoplasms with PCM1―JAK2 Myelodysplastic/myeloproliferative neoplasms Chronic myelomonocytic leukaemia Atypical chronic myeloid leukaemia, BCR-ABL1―negative Juvenile myelomonocytic leukaemia Myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis Myelodysplastic/myeloproliferative neoplasm, unclassifiable Myelodysplastic syndromes Myelodysplastic syndrome with single lineage dysplasia Myelodysplastic syndrome with ring sideroblasts and single lineage dysplasia Myelodysplastic syndrome with ring sideroblasts and multilineage dysplasia Myelodysplastic syndrome with multilineage dysplasia Myelodysplastic syndrome with excess blasts Myelodysplastic syndrome with isolated del(5q) Myelodysplastic syndrome, unclassifiable Refractory cytopenia of childhood Myeloid neoplasms with germline predisposition Acute myeloid leukaemia with germline CEBPA mutation Myeloid neoplasms with germline DDX41 mutation Myeloid neoplasms with germline RUNX1 mutation Myeloid neoplasms with germline ANKRD26 mutation Myeloid neoplasms with germline ETV6 mutation Myeloid neoplasms with germline GATA2 mutation Acute myeloid leukaemia (AML) and related precursor neoplasms AML with recurrent genetic abnormalities AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1 AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11 Acute promyelocytic leukaemia with PML-RARA AML with t(9;11)(p21.3;q23.3); KMT2A-MLLT3 AML with t(6;9)(p23;q34.1); DEK-NUP214 AML with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2, MECOM AML (megakaryoblastic) with t(1;22)(p13.3;q13.1); RBM15-MKL1 AML with BCR-ABL1 AML with mutated NPM1 AML with biallelic mutation of CEBPA AML with mutated RUNX1 AML with myelodysplasia-related changes Therapy-related myeloid neoplasms Acute myeloid leukaemia, NOS AML with minimal differentiation AML without maturation AML with maturation Acute myelomonocytic leukaemia Acute monoblastic and monocytic leukaemia Pure erythroid leukaemia Acute megakaryoblastic leukaemia Acute basophilic leukaemia Acute panmyelosis with myelofibrosis Myeloid sarcoma Myeloid proliferations associated with Down syndrome Transient abnormal myelopoiesis associated with Down syndrome Myeloid leukaemia associated with Down syndrome Blastic plasmacytoid dendritic cell neoplasm Acute leukaemias of ambiguous lineage Acute undifferentiated leukaemia Mixed-phenotype acute leukaemia with t(9;22)(q34.1;q11.2); BCR-ABL1 Mixed-phenotype acute leukaemia with t(v;11q23.3); KMT2A-rearranged Mixed-phenotype acute leukaemia, B/myeloid, NOS Mixed-phenotype acute leukaemia, T/myeloid, NOS Mixed-phenotype acute leukaemia, NOS, rare types Acute leukaemias of ambiguous lineage, NOS Lymphoid neoplasms Precursor lymphoid neoplasms B-lymphoblastic leukaemia/lymphoma, NOS B-lymphoblastic leukaemia/lymphoma with t(9;22)(q34.1;q11.2); BCR-ABL1 B-lymphoblastic leukaemia/lymphoma with t(v;11q23.3); KMT2A-rearranged B-lymphoblastic leukaemia/lymphoma with t(12;21)(p13.2;q22.1); ETV6-RUNX1 B-lymphoblastic leukaemia/lymphoma with hyperdiploidy B-lymphoblastic leukaemia/lymphoma with hypodiploidy (hypodiploid ALL) B-lymphoblastic leukaemia/lymphoma with t(5;14)(q31.1;q32.1); IGH/IL3 B-lymphoblastic leukaemia/lymphoma with t(1;19)(q23;p13.3); TCF3-PBX1 B-lymphoblastic leukaemia/lymphoma, BCR-ABL 1―like B-lymphoblastic leukaemia/lymphoma with iAMP21 T-lymphoblastic leukaemia/lymphoma Early T-cell precursor lymphoblastic leukaemia NK-lymphoblastic leukaemia/lymphoma Mature B-cell neoplasms Chronic lymphocytic leukaemia (CLL)/ small lymphocytic lymphoma Monoclonal B-cell lymphocytosis, CLL-type Monoclonal B-cell lymphocytosis, non-CLL-type B-cell prolymphocytic leukaemia Splenic marginal zone lymphoma Hairy cell leukaemia Splenic B-cell lymphoma/leukaemia, unclassifiable Splenic diffuse red pulp small B-cell lymphoma Hairy cell leukaemia variant Lymphoplasmacytic lymphoma Waldentrom macroglobulinemia IgM monoclonal gammopathy of undetermined significance Heavy chain diseases Mu heavy chain disease Gamma heavy chain disease Alpha heavy chain disease Plasma cell neoplasms Non-IgM monoclonal gammopathy of undetermined significance Plasma cell myeloma Solitary plasmacytoma of bone Extraosseous plasmacytoma Monoclonal immunoglobulin deposition diseases Primary amyloidosis Light chain and heavy chain deposition diseases Extranodal marginal zone lymphoma of mucosa- associated lymphoid tissue (MALT lymphoma) Nodal marginal zone lymphoma Paediatric nodal marginal zone lymphoma Follicular lymphoma In situ follicular neoplasia Duodenal-type follicular lymphoma Testicular follicular lymphoma Paediatric-type follicular lymphoma Large B-cell lymphoma with IRF4 rearrangement Primary cutaneous follicle centre lymphoma Mantle cell lymphoma In situ mantle cell neoplasia Diffuse large B-cell lymphoma (DLBCL), NOS Germinal centre B-cell subtype Activated B-cell subtype T-cell/histiocyte-rich large B-cell lymphoma Primary DLBCL of the CNS Primary cutaneous DLBCL, leg type EBV-positive DLBCL, NOS EBV-positive mucocutaneous ulcer DLBCL associated with chronic inflammation Fibrin-associated diffuse large B-cell lymphoma Lymphomatoid granulomatosis, grade 1,2 Lymphomatoid granulomatosis, grade 3 Primary mediastinal (thymic) large B-cell lymphoma Intravascular large B-cell lymphoma ALK-positive large B-cell lymphoma Plasmablastic lymphoma Primary effusion lymphoma Multicentric Castleman disease HHV8-positive DLBCL, NOS HHV8-positive germinotropic lymphoproliferative disorder Burkitt lymphoma Burkitt-like lymphoma with 11q aberration High-grade B-cell lymphoma High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements High-grade B-cell lymphoma, NOS B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classic Hodgkin lymphoma Mature T- and NK-cell neoplasms T-cell prolymphocytic leukaemia T-cell large granular lymphocytic leukaemia Chronic lymphoproliferative disorder of NK cells Aggressive NK-cell leukaemia Systemic EBV-positive T-cell lymphoma of childhood Chronic active EBV infection of T- and NK-cell type, systemic form Hydroa vacciniforme-like lymphoproliferative disorder Severe mosquito bite allergy Adult T-cell leukaemia/lymphoma Extranodal NK/T-cell lymphoma, nasal type Enteropathy-associated T-cell lymphoma Monomorphic epitheliotropic intestinal T-cell lymphoma Intestinal T-cell lymphoma, NOS Indolent T-cell lymphoproliferative disorder of the gastrointestinal tract Hepatosplenic T-cell lymphoma Subcutaneous panniculitis-like T-cell lymphoma Mycosis fungoides Sezary syndrome Primary cutaneous CD30-positive T-cell lymphoproliferative disorders Lymphomatoid papulosis Primary cutaneous anaplastic large cell lymphoma Primary cutaneous gamma delta T-cell lymphoma Primary cutaneous CD8-positive aggressive epidermotropic cytotoxic T-cell lymphoma Primary cutaneous acral CD8-positive T-cell lymphoma Primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder Peripheral T-cell lymphoma, NOS Angioimmunoblastic T-cell lymphoma Follicular T-cell lymphoma Nodal peripheral T-cell lymphoma with T follicular helper phenotype Anaplastic large cell lymphoma, ALK-positive Anaplastic large cell lymphoma, ALK-negative Breast implant-associated anaplastic large cell lymphoma Hodgkin lymphomas Nodular lymphocyte predominant Hodgkin lymphoma Classic Hodgkin lymphoma Nodular sclerosis classic Hodgkin lymphoma Lymphocyte-rich classic Hodgkin lymphoma Mixed cellularity classic Hodgkin lymphoma Lymphocyte-depleted classic Hodgkin lymphoma Immunodeficiency-associated lymphoproliferative disorders Post-transplant lymphoproliferative disorders (PTLD) Non-destructive PTLD Plasmacytic hyperplasia PTLD Infectious mononucleosis PTLD Florid follicular hyperplasia Polymorphic PTLD Monomorphic PTLD Classic Hodgkin Lymphoma PTLD Other iatrogenic immunodeficiency- associated lymphoproliferative disorders Histiocytic and dendritic cell neoplasms Histiocytic sarcoma Langerhans cell histiocytosis, NOS Langerhans cell histiocytosis, monostotic Langerhans cell histiocytosis, polystotic Langerhans cell histiocytosis, disseminated Langerhans cell sarcoma Indeterminate dendritic cell tumour Interdigitating dendritic cell sarcoma Follicular dendritic cell sarcoma Fibroblastic reticular cell tumour Disseminated juvenile xanthogranuloma Erdheim–Chester disease Treatment [ edit ] Treatment can occasionally consist of "watchful waiting" (e.g. in CLL ) or symptomatic treatment (e.g. blood transfusions in MDS ). ... External links [ edit ] Classification D ICD - 10 : C81 - C96 ICD - 9-CM : 200 - 209 ICD-O : 9590-9999 MeSH : D019337 v t e Leukaemias , lymphomas and related disease B cell ( lymphoma , leukemia ) (most CD19 CD20 ) By development/ marker TdT+ ALL ( Precursor B acute lymphoblastic leukemia/lymphoma ) CD5 + naive B cell ( CLL/SLL ) mantle zone ( Mantle cell ) CD22 + Prolymphocytic CD11c+ ( Hairy cell leukemia ) CD79a + germinal center / follicular B cell ( Follicular Burkitt's GCB DLBCL Primary cutaneous follicle center lymphoma ) marginal zone / marginal zone B-cell ( Splenic marginal zone MALT Nodal marginal zone Primary cutaneous marginal zone lymphoma ) RS ( CD15 +, CD30 +) Classic Hodgkin lymphoma ( Nodular sclerosis ) CD20+ ( Nodular lymphocyte predominant Hodgkin lymphoma ) PCDs / PP ( CD38 +/ CD138 +) see immunoproliferative immunoglobulin disorders By infection KSHV ( Primary effusion ) EBV Lymphomatoid granulomatosis Post-transplant lymphoproliferative disorder Classic Hodgkin lymphoma Burkitt's lymphoma HCV Splenic marginal zone lymphoma HIV ( AIDS-related lymphoma ) Helicobacter pylori ( MALT lymphoma ) Cutaneous Diffuse large B-cell lymphoma Intravascular large B-cell lymphoma Primary cutaneous marginal zone lymphoma Primary cutaneous immunocytoma Plasmacytoma Plasmacytosis Primary cutaneous follicle center lymphoma T/NK T cell ( lymphoma , leukemia ) (most CD3 CD4 CD8 ) By development/ marker TdT+ : ALL ( Precursor T acute lymphoblastic leukemia/lymphoma ) prolymphocyte ( Prolymphocytic ) CD30+ ( Anaplastic large-cell lymphoma Lymphomatoid papulosis type A ) Cutaneous MF+variants indolent: Mycosis fungoides Pagetoid reticulosis Granulomatous slack skin aggressive: Sézary disease Adult T-cell leukemia/lymphoma Non-MF CD30 -: Non-mycosis fungoides CD30− cutaneous large T-cell lymphoma Pleomorphic T-cell lymphoma Lymphomatoid papulosis type B CD30 +: CD30+ cutaneous T-cell lymphoma Secondary cutaneous CD30+ large-cell lymphoma Lymphomatoid papulosis type A Other peripheral Hepatosplenic Angioimmunoblastic Enteropathy-associated T-cell lymphoma Peripheral T-cell lymphoma not otherwise specified ( Lennert lymphoma ) Subcutaneous T-cell lymphoma By infection HTLV-1 ( Adult T-cell leukemia/lymphoma ) NK cell / (most CD56 ) Aggressive NK-cell leukemia Blastic NK cell lymphoma T or NK EBV ( Extranodal NK-T-cell lymphoma / Angiocentric lymphoma ) Large granular lymphocytic leukemia Lymphoid+ myeloid Acute biphenotypic leukaemia Lymphocytosis Lymphoproliferative disorders ( X-linked lymphoproliferative disease Autoimmune lymphoproliferative syndrome ) Leukemoid reaction Diffuse infiltrative lymphocytosis syndrome Cutaneous lymphoid hyperplasia Cutaneous lymphoid hyperplasia with bandlike and perivascular patterns with nodular pattern Jessner lymphocytic infiltrate of the skin General Hematological malignancy leukemia Lymphoproliferative disorders Lymphoid leukemias v t e Myeloid -related hematological malignancy CFU-GM / and other granulocytes CFU-GM Myelocyte AML : Acute myeloblastic leukemia M0 M1 M2 APL/M3 MP Chronic neutrophilic leukemia Monocyte AML AMoL/M5 Myeloid dendritic cell leukemia CML Philadelphia chromosome Accelerated phase chronic myelogenous leukemia Myelomonocyte AML M4 MD-MP Juvenile myelomonocytic leukemia Chronic myelomonocytic leukemia Other Histiocytosis CFU-Baso AML Acute basophilic CFU-Eos AML Acute eosinophilic MP Chronic eosinophilic leukemia / Hypereosinophilic syndrome MEP CFU-Meg MP Essential thrombocytosis Acute megakaryoblastic leukemia CFU-E AML Erythroleukemia/M6 MP Polycythemia vera MD Refractory anemia Refractory anemia with excess of blasts Chromosome 5q deletion syndrome Sideroblastic anemia Paroxysmal nocturnal hemoglobinuria Refractory cytopenia with multilineage dysplasia CFU-Mast Mastocytoma Mast cell leukemia Mast cell sarcoma Systemic mastocytosis Mastocytosis : Diffuse cutaneous mastocytosis Erythrodermic mastocytosis Adult type of generalized eruption of cutaneous mastocytosis Urticaria pigmentosa Mast cell sarcoma Solitary mastocytoma Systemic mastocytosis Xanthelasmoidal mastocytosis Multiple/unknown AML Acute panmyelosis with myelofibrosis Myeloid sarcoma MP Myelofibrosis Acute biphenotypic leukaemia
-
Tourette Syndrome
Wikipedia
Neurodevelopmental disorder involving motor and vocal tics Tourette syndrome Other names Tourette's syndrome, Tourette's disorder, Gilles de la Tourette syndrome (GTS), combined vocal and multiple motor tic disorder [de la Tourette] Georges Gilles de la Tourette (1857–1904), namesake of Tourette syndrome Specialty Pediatrics , neurology Symptoms Tics [1] Usual onset Typically in childhood [1] Duration Long term [2] Causes Genetic with environmental influence [2] Diagnostic method Based on history and symptoms [1] Medication Usually none, occasionally neuroleptics and noradrenergics [1] Prognosis Improvement to disappearance of tics beginning in late teens [2] Frequency About 1% [3] Tourette syndrome or Tourette's syndrome (abbreviated as TS or Tourette's ) is a common neurodevelopmental disorder that begins in childhood or adolescence. ... Tourette's was once regarded as a rare and bizarre syndrome and has popularly been associated with coprolalia (the utterance of obscene words or socially inappropriate and derogatory remarks). ... There is also a broader hypothesis that links immune-system abnormalities and immune dysregulation with TS. [10] [36] Some forms of OCD may be genetically linked to Tourette's, [29] although the genetic factors in OCD with and without tics may differ. [9] The genetic relationship of ADHD to Tourette syndrome, however, has not been fully established. [55] [66] [46] A genetic link between autism and Tourette's has not been established as of 2017. [41] Mechanism The basal ganglia and thalamus are implicated in Tourette syndrome. ... [b] Tourette syndrome was once thought to be rare: in 1972, the US National Institutes of Health (NIH) believed there were fewer than 100 cases in the United States, [119] and a 1973 registry reported only 485 cases worldwide. [120] However, numerous studies published since 2000 have consistently demonstrated that the prevalence is much higher. [121] Recognizing that tics may often be undiagnosed and hard to detect, [c] newer studies use direct classroom observation and multiple informants (parents, teachers and trained observers), and therefore record more cases than older studies. [90] [124] As the diagnostic threshold and assessment methodology have moved towards recognition of milder cases, the estimated prevalence has increased. [121] History Main article: History of Tourette syndrome Jean-Martin Charcot was a French neurologist and professor who named Tourette syndrome for his intern, Georges Gilles de la Tourette. ... Not everyone with Tourette's wants treatment or a cure, especially if that means they may lose something else in the process. [93] [138] The researchers Leckman and Cohen believe that there may be latent advantages associated with an individual's genetic vulnerability to developing Tourette syndrome that may have adaptive value, such as heightened awareness and increased attention to detail and surroundings. [139] [140] Accomplished musicians, athletes, public speakers and professionals from all walks of life are found among people with Tourette's. [77] [141] The athlete Tim Howard , described by the Chicago Tribune as the "rarest of creatures—an American soccer hero", [142] and by the Tourette Syndrome Association as the "most notable individual with Tourette Syndrome around the world", [143] says that his neurological makeup gave him an enhanced perception and an acute focus that contributed to his success on the field. [110] Samuel Johnson is a historical figure who likely had Tourette syndrome, as evidenced by the writings of his friend James Boswell . [144] [145] Johnson wrote A Dictionary of the English Language in 1747, and was a prolific writer, poet, and critic.

-
Chronic Fatigue Syndrome
Wikipedia
Medical condition involving extreme fatigue among other symptoms Chronic fatigue syndrome Other names Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS), [1] myalgic encephalomyelitis (ME), post-viral fatigue syndrome (PVFS), chronic fatigue immune dysfunction syndrome (CFIDS), systemic exertion intolerance disease (SEID), others [2] : 20 Specialty Primary care , neurology , rheumatology , infectious diseases , physical therapy , occupational therapy , mental health , behavioral health [2] : 223 Symptoms Worsening of symptoms with activity , long-term fatigue , others [1] Usual onset 40 to 60 years old [3] Duration Often years [4] Causes Unknown [1] Risk factors Female gender, virus and bacterial infections, genetics , major injury , bodily response to severe stress and others [5] [6] : 1–2 Diagnostic method Based on symptoms [1] Treatment Symptomatic [7] [8] Frequency About 0.68 to 1% globally [9] [10] Chronic fatigue syndrome ( CFS ), also called myalgic encephalomyelitis ( ME ) and ME/CFS , is a complex, fatiguing, long-term medical condition diagnosed by required primary symptoms and criteria, often involving a broad range of symptoms. ... In many chronic illnesses, aerobic exercise is beneficial, but in chronic fatigue syndrome, the CDC does not recommend it. ... An initial link to the Epstein-Barr virus had the illness acquire the name "chronic Epstein-Barr virus syndrome". [2] : 29 [87] In 1987, the CDC convened a working group to reach a consensus on the clinical features of the illness. The working group concluded that CFS was not new, and that the many different names given to it previously reflected widely differing concepts of the illness's cause and epidemiology. [146] The CDC working group chose "chronic fatigue syndrome" as a more neutral and inclusive name for the illness, but noted that "myalgic encephalomyelitis" was widely accepted in other parts of the world. [87] In 1988, the first definition of CFS was published. ... Currently, the most commonly used are "chronic fatigue syndrome", "myalgic encephalomyelitis", and the umbrella term "ME/CFS".FURIN, NR3C1, RNASEL, NTRK3, ETV6, TNF, TRPM3, IL10, COMT, SLC6A4, TGFB1, NHS, NOS2, TPH2, HTR2A, APRT, MFAP1, HLA-DRB1, PTGS2, EIF2AK2, SERPINA6, HPSE, MTA2, MASP2, GDF15, ADIPOQ, ASIC3, TRPA1, TRBV20OR9-2, VIP, TRPM2, PLAU, SULT1E1, TH, PRKCD, REN, ACTB, TMED2, TPPP, ERVK-18, CD24, MIR330, MIR99B, MIR30C2, MIR30C1, MIR150, MIR143, MIR126, COPD, EMX2OS, NRSN1, EMB, RBM45, NLRP3, IL17F, SIGLEC12, FANCM, KRT20, REM1, SESN1, DISC1, B3GAT1, CHP1, PSIP1, PF4, NFE2L2, OAS2, NPAS2, GABPA, FLNA, FCN1, PTK2B, ESR2, ELANE, DUT, ACE, DBI, CTSB, CPT2, MAP3K8, CD69, CD38, MS4A1, CD14, CD9, CD6, BLM, CFB, AVP, ATR, AMPD1, GCH1, GDNF, GRIK2, IL2, ADCYAP1, MYOG, MUC1, RNR2, MBL2, LTA, KLRC2, KIR3DS1, IL18, IL6, IL1B, HTT, IL1A, IGHG3, IFNB1, IFNA13, IFNA1, HTR7, HTR1A, HLA-DQB1, HLA-DQA1, CFH, H3P19
-
Hydrocephalus With Stenosis Of The Aqueduct Of Sylvius
Orphanet
A congenital, X-linked, clinical subtype of L1 syndrome characterized by severe hydrocephalus often of prenatal onset, adducted thumbs, spasticity (mostly evidenced by brisk tendon reflexes and extensor plantar responses) and moderate to severe intellectual disability. This subtype represents the severe end of the L1 syndrome spectrum and is associated with poor prognosis.
-
3-Phosphoserine Phosphatase Deficiency, Infantile/juvenile Form
Orphanet
3-Phosphoserine phosphatase deficiency is an extremely rare form of serine deficiency syndrome (see this term) characterized clinically by congenital microcephaly and severe psychomotor retardation in the single reported case to date, which was associated with Williams syndrome (see this term).





