Differential diagnosis The differential diagnoses are those diseases that produce white dots in choroid and retina; they include white dot syndromes, vogt-Koyanagi-Harada disease, infectious etiologies (Lyme disease, tuberculosis, toxoplasmosis) and primary intraocular lymphoma (masquerade syndromes) (see these terms).
These depigmented spots, the most distinctive sign of the syndrome, appear at the level of the retinal pigment epithelium but, on occasion, suggest an even deeper infiltration.
Birdshot chorioretinopathy is an eye condition in which painless, light-colored spots develop on the retina . These spots are scattered in a "birdshot" pattern. The effects of this condition on vision are quite variable; some individuals' vision is only mildly affected, whereas others experience a significant decline in vision, the appearance of floaters (small specks that appear in one's line of sight), night blindness , and other vision problems. Symptoms typically begin around middle age; Caucasians are affected more than individuals of other ethnicities. The cause of birdshot chorioretinopathy is currently unknown, but it is suspected to be an autoimmune disease. Treatment may include medications that aim to regulate the body's immune response .
In the presence of anosmia, idiopathic hypogonadotropic hypogonadism has been called 'Kallmann syndrome (KS),' whereas in the presence of a normal sense of smell, it has been termed 'normosmic idiopathic hypogonadotropic hypogonadism (nIHH)' (summary by Raivio et al., 2007). ... They also stated that luteinizing hormone-releasing hormone (LRH) treatment may be helpful; that associated hypothalamic-pituitary-prolactin dysfunction may be present; and that FIGD and the Kallmann syndrome (see 147950) are distinct entities. To further define the genetic and phenotypic variability of FIGD, Waldstreicher et al. (1996) reviewed detailed family histories of 106 cases of GNRH deficiency with or without anosmia, i.e., Kallmann syndrome or idiopathic hypogonadotropic hypogonadism (IHH). ... However, of the families in which the proband was the sole member affected by Kallmann syndrome or IHH, 9 had individuals with isolated anosmia, and 8 had a strong history of delayed puberty. If these phenotypes were considered as variable expressions of Kallmann syndrome or IHH seen in the proband, then 34% of the cases could be considered familial.
NPHP3 mutations can also result in the more severe disorder Meckel syndrome-7 (MKS7; 227010). Genetic Heterogeneity of Renal-Hepatic-Pancreatic Dysplasia See also RHPD2 (615415), caused by mutation in the NEK8 gene (609799) on chromosome 17q11. ... However, Bernstein et al. (1987) noted that similar renal, hepatic, and pancreatic abnormalities appear in other syndromes, including trisomy 9, Meckel syndrome (see 249000), the chondrodysplasias of Jeune syndrome and Saldino and Noonan syndrome (see 613091), and type II glutaric acidemia (231680). The authors concluded that after exclusion of identifiable syndromes, the remaining cases of renal-hepatic-pancreatic dysplasia do not necessarily constitute a homogeneous group.
A number sign (#) is used with this entry because of evidence that renal-hepatic-pancreatic dysplasia-2 (RHPD2) is caused by homozygous or compound heterozygous mutation in the NEK8 gene (609799) on chromosome 17q11. Description RHPD2 is an autosomal recessive multisystemic disorder with severe abnormalities apparent in utero and often resulting in fetal death or death in infancy. The main organs affected include the kidney, liver, and pancreas, although other abnormalities, including cardiac, skeletal, and lung defects, may also be present. Affected individuals often have situs inversus. The disorder results from a defect in ciliogenesis and ciliary function, as well as in cell proliferation and epithelial morphogenesis; thus, the clinical manifestations are highly variable (summary by Grampa et al., 2016). For a discussion of genetic heterogeneity of renal-hepatic-pancreatic dysplasia, see RHPD1 (208540).
Renal-hepatic-pancreatic dysplasia is a rare, genetic, developmental defect during embryogenesis syndrome characterized by the triad of pancreatic fibrosis (and cysts, with a reduction of parenchymal tissue), renal dysplasia (with peripheral cortical cysts, primitive collecting ducts, glomerular cysts and metaplastic cartilage) and hepatic dysgenesis (enlarged portal areas containing numerous elongated binary profiles with a tendancy to perilobular fibrosis).
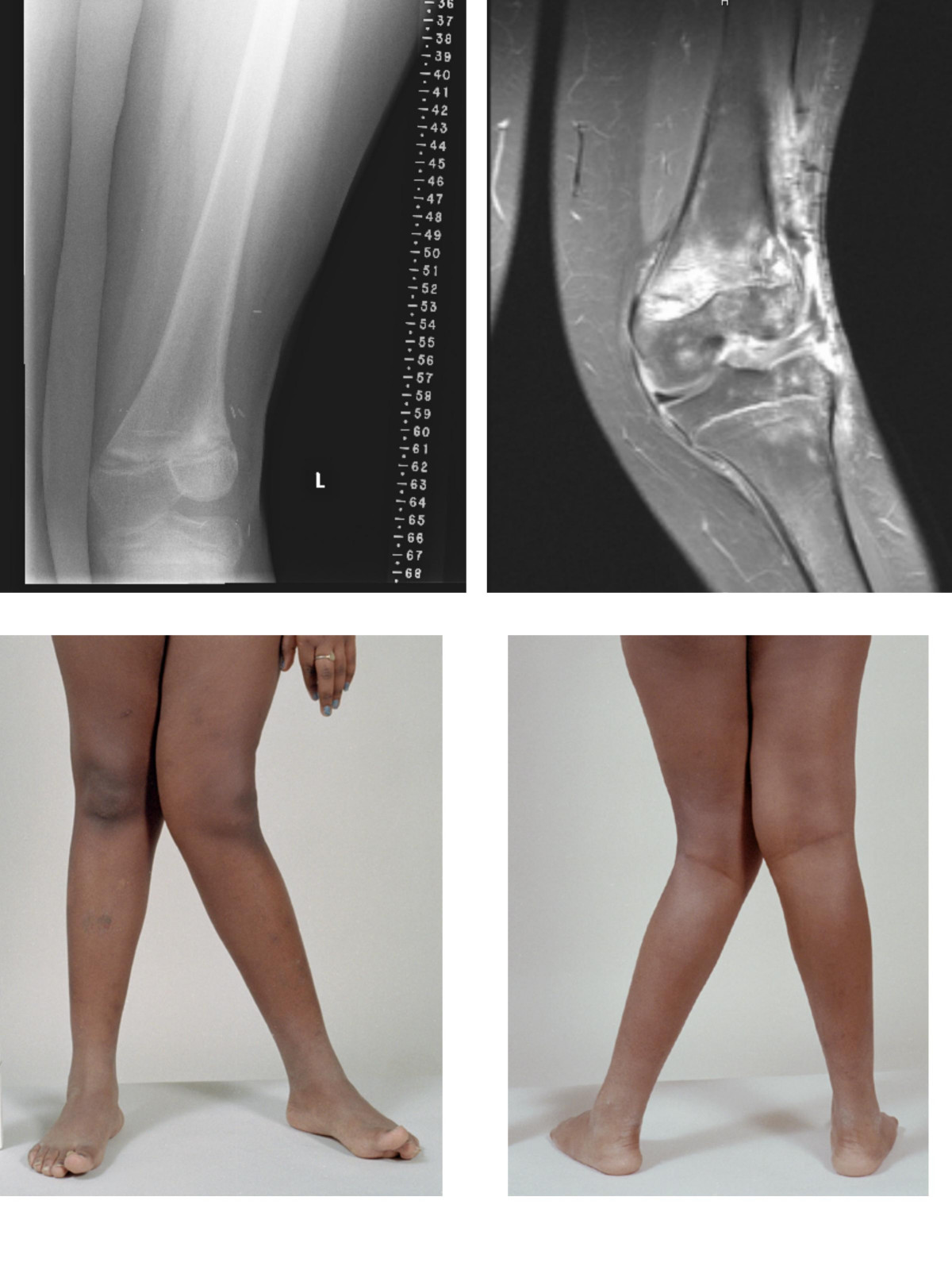
Human disease Patellar tendinitis Other names Quadriceps tendinopathy, patellar tendinopathy, jumper's knee, patellar tendinosis, patellar tendinitis Location of the pain in patellar tendinitis Specialty Orthopedics , sports medicine Symptoms Pain at the front of the knee [1] Complications Patellar tendon rupture [2] Risk factors Jumping sports, being overweight [1] Diagnostic method Based on symptoms and examination [2] Differential diagnosis Chondromalacia patella , Osgood-Schlatter disease , patellofemoral syndrome , infrapatellar bursitis [1] [2] Treatment Rest, physical therapy [2] Prognosis Recovery can be slow [2] Frequency 14% of athletes [1] Patellar tendinitis , also known as jumper's knee , is an overuse injury of the tendon that straightens the knee . [1] Symptoms include pain in the front of the knee. [1] Typically the pain and tenderness is at the lower part of the kneecap , though the upper part may also be affected. [2] Generally there is not pain when the person is at rest. [2] Complications may include patellar tendon rupture . [2] Risk factors include being involved in athletics and being overweight . [1] It is particularly common in athletes who are involved in jumping sports such as basketball and volleyball. [1] [2] The underlying mechanism involves small tears in the tendon connecting the kneecap with the shinbone . [2] Diagnosis is generally based on symptoms and examination . [2] Other conditions that can appear similar include infrapatellar bursitis , chondromalacia patella and patellofemoral syndrome . [1] [2] Treatment often involves resting the knee and physical therapy . [2] Evidence for treatments, including rest, however is poor. [3] [4] Recovery can take a year. [2] It is relatively common with about 14% of athletes currently affected. [1] Males are more commonly affected than females. [2] The term "jumper's knee" was coined in 1973. [2] Contents 1 Signs and symptoms 2 Causes 3 Diagnosis 4 Treatment 4.1 Procedures 4.2 Surgery 5 Epidemiology 6 References 7 External links Signs and symptoms [ edit ] People report anterior knee pain, often with an aching quality. ... External links [ edit ] Classification D ICD - 10 : M76.5 ICD - 9-CM : 726.64 DiseasesDB : 9704 External resources eMedicine : sports/56 v t e Soft tissue disorders Capsular joint Synoviopathy Synovitis / Tenosynovitis Calcific tendinitis Stenosing tenosynovitis Trigger finger De Quervain syndrome Transient synovitis Ganglion cyst osteochondromatosis Synovial osteochondromatosis Plica syndrome villonodular synovitis Giant-cell tumor of the tendon sheath Bursopathy Bursitis Olecranon Prepatellar Trochanteric Subacromial Achilles Retrocalcaneal Ischial Iliopsoas Synovial cyst Baker's cyst Calcific bursitis Noncapsular joint Symptoms Ligamentous laxity Hypermobility Enthesopathy / Enthesitis / Tendinopathy upper limb Adhesive capsulitis of shoulder Impingement syndrome Rotator cuff tear Golfer's elbow Tennis elbow lower limb Iliotibial band syndrome Patellar tendinitis Achilles tendinitis Calcaneal spur Metatarsalgia Bone spur other/general: Tendinitis / Tendinosis Nonjoint Fasciopathy Fasciitis : Plantar Nodular Necrotizing Eosinophilic Fibromatosis / contracture Dupuytren's contracture Plantar fibromatosis Aggressive fibromatosis Knuckle pads
Overview Patellar tendinitis is an injury to the tendon connecting your kneecap (patella) to your shinbone. The patellar tendon works with the muscles at the front of your thigh to extend your knee so that you can kick, run and jump. Patellar tendinitis, also known as jumper's knee, is most common in athletes whose sports involve frequent jumping — such as basketball and volleyball. However, even people who don't participate in jumping sports can get patellar tendinitis. For most people, treatment of patellar tendinitis begins with physical therapy to stretch and strengthen the muscles around the knee.
Balkan endemic nephropathy is a kidney disease that affects people living in rural areas of Bosnia, Bulgaria, Croatia, Romania, and Serbia (areas along the Danube river and its tributaries). Affected individuals develop kidney damage that slowly progresses over 10 to 20 years to kidney failure . Many people with this condition also develop a type of bladder cancer known as upper urothelial carcinoma (UUC). Balkan endemic nephropathy is caused by chronic dietary exposure to low concentrations of a toxin called aristolochic acid , which comes from a plant called Aristolochia clematis . Genetics factors may also be involved. There is no specific prevention or treatment of this condition.
Primary failure of eruption has been known to be associated with Parathyroid hormone 1 receptor mutation. [1] Contents 1 Primary failure of eruption 1.1 Management 2 Mechanical failure of eruption 3 Eruption failure related to syndrome 4 References Primary failure of eruption [ edit ] The term primary failure of eruption was named by William Proffit and Katherine Vig in 1981. [2] [3] This type of failure of eruption has a genetic or familial background precursor as a cause. ... In mechanical failure of eruption, affected tooth has partial or complete loss of PDL in a panoramic radiograph and teeth distal to affected tooth do not have this condition. [6] [7] On a percussion test, a tooth with mechanical failure of eruption will have a dull metallic sound. Eruption failure related to syndrome [ edit ] There have been many syndromes which have been identified to be related to failure of eruption of teeth. These syndromes are Cleidocranial dyspalsia , Osteoporosis , Rutherford syndrome , GAPO syndrome and Osteoglophonic dysplasia .
A number sign (#) is used with this entry because of evidence that primary failure of tooth eruption (PFE) is caused by heterozygous mutation in the PTHR1 gene (168468) on chromosome 3p21. See also 157950 and 273050 for phenotypes with shared features of PFE. Clinical Features Shokeir (1974) described autosomal dominant inheritance of failure of eruption of permanent teeth. The primary dentition persisted in the adult; however, the proband showed complete or partial eruption of 11 permanent teeth. Proffit and Vig (1981) described the characteristic features of primary failure of tooth eruption: posterior teeth are more commonly affected, and the bite distal to the first affected tooth is usually completely open; the affected teeth may or may not have initially erupted into occlusion before submerging; deciduous teeth, especially second deciduous molars are commonly submerged; involvement may be unilateral or bilateral; involved permanent teeth may become ankylosed after failure of eruption has occurred; orthodontic extrusion is unsuccessful and usually leads to ankylosis; and although other members of the family may be affected they are not usually close relatives.
A rare genetic odontologic disease characterized by failure of eruption of non-ankylosed permanent teeth without evidence of obvious mechanical obstruction. Posterior teeth are preferentially affected (typically with involvement of all teeth distal to the most mesial non-erupted tooth), resulting in a posterior open bite. Non-ankylosed teeth tend to become ankylosed, and orthodontic treatment of affected teeth is generally unsuccessful.
The fetus is said to suffer from acrania if it meets the following criteria: the foetus should have a perfectly normal facial bone, a normal cervical column but without the fetal skull and a volume of brain tissue equivalent to at least one third of the normal brain size. [2] Contents 1 Causes 1.1 Genetics 1.1.1 Genetic counseling 1.2 Amniotic band syndrome 2 Mechanism 2.1 Ectodermal mesenchyme 3 Diagnosis 4 Prognosis 5 References 6 External links Causes [ edit ] Infant with both acrania and anencephaly . ... In order to make genetic counseling for families easier this disease is often differentially diagnosed with other diseases that can occur at the same time such as anencephaly and acalvaria , though these diseases may not always occur simultaneously. [1] While this disease is tragic, reoccurrence rates are extremely low so genetic counseling is not always necessary. [1] Amniotic band syndrome [ edit ] During amniotic band syndrome (ABS), fibrous bands may entrap various parts of the developing fetus causing malformations. ... "Prenatal Diagnosis of Acrania Associated with Amniotic Band Syndrome". Obstetrics & Gynecology . Elsevier. 102 (5 (part 2)): 1176–1178. doi : 10.1016/S0029-7844(03)00118-2 . ... External links [ edit ] Classification D ICD - 10 : Q00.0 ICD - 9-CM : 740.0 SNOMED CT : 203923004 External resources Orphanet : 945 Acalvaria (Acrania) at NIH 's Office of Rare Diseases v t e Congenital malformations and deformations of nervous system Brain Neural tube defect Anencephaly Acephaly Acrania Acalvaria Iniencephaly Encephalocele Chiari malformation Other Microcephaly Congenital hydrocephalus Dandy–Walker syndrome other reduction deformities Holoprosencephaly Lissencephaly Microlissencephaly Pachygyria Hydranencephaly Septo-optic dysplasia Megalencephaly Hemimegalencephaly CNS cyst Porencephaly Schizencephaly Polymicrogyria Bilateral frontoparietal polymicrogyria Spinal cord Neural tube defect Spina bifida Rachischisis Other Currarino syndrome Diastomatomyelia Syringomyelia
Primary acalvaria is an extremely rare malformation characterized by the absence of the flat skull bones of the brain, dura mater , and scalp muscles. The skull base and facial features are fully formed and usually appear normal. The cause of acalvaria is still unknown. Acalvaria can be distinguished from anencephaly , the most common differential diagnosis, by the presence of a layer of skin overlying the brain matter and normal cerebral hemispheres. This malformation is most often lethal at birth due to other associated anomalies or to trauma during delivery, but a few surviving infants have been reported. Prenatal diagnosis via transvaginal ultrasound and/or magnetic resonance imaging is critical for better pregnancy management.
A rare malformation characterized by missing scalp and flat bones over an area of the cranial vault. The size of the affected area is variable. In rare cases, acalvaria involves the whole of the dome-like superior portion of the cranium comprising the frontal, parietal, and occipital bones. Dura mater and associated muscles are absent in the affected area but the central nervous system is usually unaffected, although some neuropathological abnormality is often present ( e.g. holoprosencephaly or gyration anomalies). Skull base and facial bones are normal. Epidemiology The prevalence is less than 1 per 100,000 births. Etiology Acalvaria is regarded as a postneurulation defect. The presumed pathogenesis of acalvaria is faulty migration of the membranous neurocranium with normal placement of the embryonic ectoderm, resulting in absence of the calvaria but an intact layer of skin over the brain parenchyma.
The skin lacks elastic recoil, in marked contrast to the hyperelasticity apparent in classic Ehlers-Danlos syndrome (see 130000). These properties are nearly always attributable to loss, fragmentation, or severe disorganization of dermal elastic fibers (summary by Davidson and Giro, 2002). ... Clinical Features Urban et al. (2009) described 4 unrelated patients with a syndrome that disrupted pulmonary, gastrointestinal, urinary, musculoskeletal, craniofacial, and dermal development. ... Urban et al. (2009) proposed the name Urban-Rifkin-Davis syndrome (URDS) for this disorder. Callewaert et al. (2013) studied affected individuals from 9 families with cutis laxa and mutations in the LTB4 gene (see MOLECULAR GENETICS). ... They also suggested that given the potential similarities in disease mechanisms between this syndrome and the Marfan syndrome (154700), pharmacologic intervention for normalization of TGF-beta signaling may be a treatment option.