Granuloma annulare (GA) is skin disorder that most often causes a rash with red bumps (erythematous papules) arranged in a circle or ring pattern (annular). GA is not contagious and is not cancerous. The rash may be localized or generalized. Localized GA is the most common form of GA (75% of the cases) and usually affects the forearms, hands, or feet. The generalized form of GA (15% of cases) presents with numerous erythematous papules that form larger, slightly raised patches (plaques) anywhere on the body, including the palms of hands and soles of feet. The plaques may or may not be in the ring pattern and can vary in color.
Overview Granuloma annulare (gran-u-LOW-muh an-u-LAR-e) is a skin condition that causes a raised rash or bumps in a ring pattern. The most common type affects young adults, usually on the hands and feet. Minor skin injuries and some medicines might trigger the condition. It's not contagious and usually not painful, but it can make you feel self-conscious. And if it becomes a long-term condition, it can cause emotional distress. Treatment might clear the skin gradually, but the bumps tend to come back.
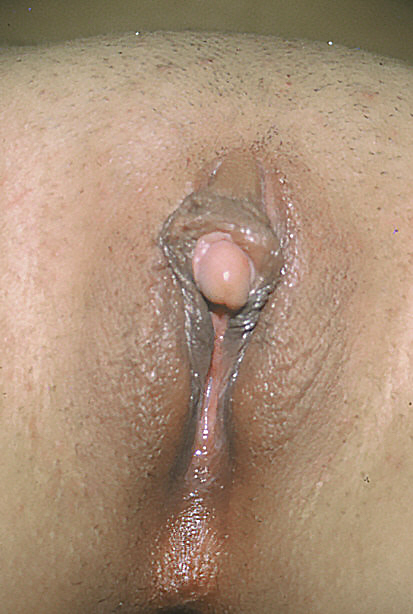
It can also be caused by the autosomal recessive congenital disorder known as Fraser syndrome . [9] In acquired clitoromegaly, the main cause is endocrine hormonal imbalance affecting the adult woman, including polycystic ovarian syndrome (PCOS) [10] and hyperthecosis . ... "Der genitalbefund beim Pseudohermaproditismus femininus des kongenitalen adrenogenitalen Syndroms. Morphologie, Hausfigkeit, Entwicklung und Vererbung der verschiedenen Genitalformen" [Genital findings in the female pseudo-hermaphroditism of the congenital adrenogenital syndrome; morphology, frequency, development and heredity of the different genital forms]. ... S2CID 19741452 . ^ van Haelst MM, Scambler PJ, Hennekam RC (December 2007). "Fraser syndrome: a clinical study of 59 cases and evaluation of diagnostic criteria". ... S2CID 25053508 . ^ Mukhtar I Khan, MD. "Polycystic Ovarian Syndrome" . Retrieved 2008-09-28 . ^ Horejsí J (June 1997).
. ^ Varki, Ajit (15 September 2007). "Trousseau's syndrome: multiple definitions and multiple mechanisms" . ... External links [ edit ] Classification D ICD - 10 : I80 , I82.1 ICD - 9-CM : 451 MeSH : D013924 SNOMED CT : 64156001 External resources MedlinePlus : 001108 Scholia has a topic profile for Thrombophlebitis . v t e Cardiovascular disease (vessels) Arteries , arterioles and capillaries Inflammation Arteritis Aortitis Buerger's disease Peripheral artery disease Arteriosclerosis Atherosclerosis Foam cell Fatty streak Atheroma Intermittent claudication Critical limb ischemia Monckeberg's arteriosclerosis Arteriolosclerosis Hyaline Hyperplastic Cholesterol LDL Oxycholesterol Trans fat Stenosis Carotid artery stenosis Renal artery stenosis Other Aortoiliac occlusive disease Degos disease Erythromelalgia Fibromuscular dysplasia Raynaud's phenomenon Aneurysm / dissection / pseudoaneurysm torso : Aortic aneurysm Abdominal aortic aneurysm Thoracic aortic aneurysm Aneurysm of sinus of Valsalva Aortic dissection Aortic rupture Coronary artery aneurysm head / neck Intracranial aneurysm Intracranial berry aneurysm Carotid artery dissection Vertebral artery dissection Familial aortic dissection Vascular malformation Arteriovenous fistula Arteriovenous malformation Telangiectasia Hereditary hemorrhagic telangiectasia Vascular nevus Cherry hemangioma Halo nevus Spider angioma Veins Inflammation Phlebitis Venous thrombosis / Thrombophlebitis primarily lower limb Deep vein thrombosis abdomen Hepatic veno-occlusive disease Budd–Chiari syndrome May–Thurner syndrome Portal vein thrombosis Renal vein thrombosis upper limb / torso Mondor's disease Paget–Schroetter disease head Cerebral venous sinus thrombosis Post-thrombotic syndrome Varicose veins Gastric varices Portacaval anastomosis Caput medusae Esophageal varices Hemorrhoid Varicocele Other Chronic venous insufficiency Chronic cerebrospinal venous insufficiency Superior vena cava syndrome Inferior vena cava syndrome Venous ulcer Arteries or veins Angiopathy Macroangiopathy Microangiopathy Embolism Pulmonary embolism Cholesterol embolism Paradoxical embolism Thrombosis Vasculitis Blood pressure Hypertension Hypertensive heart disease Hypertensive emergency Hypertensive nephropathy Essential hypertension Secondary hypertension Renovascular hypertension Benign hypertension Pulmonary hypertension Systolic hypertension White coat hypertension Hypotension Orthostatic hypotension v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline Authority control NDL : 01195329
Lasting leg pain and swelling (post-phlebetic syndrome). This condition, also known as post-thrombotic syndrome, can develop months or years after you've had DVT .
Overview Neurodermatitis is a skin condition that starts with an itchy patch of skin. Scratching makes it itch more. With more scratching, the skin becomes thick and leathery. You may develop several itchy spots, typically on the neck, wrists, forearms, legs or groin area. Neurodermatitis — also known as lichen simplex chronicus — is not life-threatening or contagious. But the itching can be so intense it disrupts your sleep, sexual function and quality of life.
It is classified under "M4" in the French-American-British classification (FAB). [1] It is classified under "AML, not otherwise classified" in the WHO classification. [2] Translocations have been observed. [3] Progression from myelodysplastic syndrome has been reported. [4] Contents 1 Signs and Symptoms 2 Cause 3 Mechanism 4 Diagnosis 5 Treatment 6 Prognosis 7 Epidemiology 8 Research Directions 9 See also 10 References 11 External links Signs and Symptoms [ edit ] Some patients may experience: [5] Fatigue Easy Bruising Abnormal Bleeding Anemia Thrombocytopenia Dyspnea If the blast count gets too high and clog up blood vessels, some patients may experience: [6] Slurred Speech Headache Confusion Weakness on one side of the body Sleepiness Cause [ edit ] The cause has not yet been determined. ... Those with AML-M4 inv(16) have a favorable prognosis with a five year overall survival rate of 61%. [12] Epidemiology [ edit ] AML is commonly seen in pediatric patients with higher pediatric incidence in Hispanics and Asians as compared to non-Hispanic Caucasian and African Americans in the USA. [12] Predisposition to AML includes but not limited to: Down syndrome , Klinefelter's syndrome , and Fanconi's anemia . [12] Acquired predisposing factors include: Aplastic anemia , Chemotherapy, prenatal exposure to tobacco, marihuana, and alcohol. [12] Research Directions [ edit ] Considering the disease is rare, not much research is being done specifically for the AML-M4 subtype. ... "Rare t(1;11)(q23;p15) in therapy-related myelodysplastic syndrome evolving into acute myelomonocytic leukemia: a case report and review of the literature". ... External links [ edit ] Classification D ICD - 10-CM : C92.5 ICD-O : M9867/3 MeSH : D015479 External resources Orphanet : 517 v t e Myeloid -related hematological malignancy CFU-GM / and other granulocytes CFU-GM Myelocyte AML : Acute myeloblastic leukemia M0 M1 M2 APL/M3 MP Chronic neutrophilic leukemia Monocyte AML AMoL/M5 Myeloid dendritic cell leukemia CML Philadelphia chromosome Accelerated phase chronic myelogenous leukemia Myelomonocyte AML M4 MD-MP Juvenile myelomonocytic leukemia Chronic myelomonocytic leukemia Other Histiocytosis CFU-Baso AML Acute basophilic CFU-Eos AML Acute eosinophilic MP Chronic eosinophilic leukemia / Hypereosinophilic syndrome MEP CFU-Meg MP Essential thrombocytosis Acute megakaryoblastic leukemia CFU-E AML Erythroleukemia/M6 MP Polycythemia vera MD Refractory anemia Refractory anemia with excess of blasts Chromosome 5q deletion syndrome Sideroblastic anemia Paroxysmal nocturnal hemoglobinuria Refractory cytopenia with multilineage dysplasia CFU-Mast Mastocytoma Mast cell leukemia Mast cell sarcoma Systemic mastocytosis Mastocytosis : Diffuse cutaneous mastocytosis Erythrodermic mastocytosis Adult type of generalized eruption of cutaneous mastocytosis Urticaria pigmentosa Mast cell sarcoma Solitary mastocytoma Systemic mastocytosis Xanthelasmoidal mastocytosis Multiple/unknown AML Acute panmyelosis with myelofibrosis Myeloid sarcoma MP Myelofibrosis Acute biphenotypic leukaemia
A rare acute myeloid leukemia disorder characterized by increased blast cells (myeloblasts, monoblast, and/or promonoblasts), representing more than 20% of the total bone marrow (BM) or peripheral blood differential counts, with 20-80% of BM cells being of monocytic lineage. Clinical presentation is the result of bone marrow involvement and extramedullary infiltration by the leukemic cells and includes asthenia, pallor, fever, dizziness, respiratory symptoms, easy bruising, bleeding disorders, and neurological deficits. Gingival hyperplasia, organomegaly, especially hepatosplenomegaly, and lymphadenopathy may also be associated.
Acute myelomonocytic leukemia (AMML) is a cancer that typically develops in the bone marrow and blood of older individuals. AMML is one type of acute myeloid leukemia , a group of blood cancers that occur when the amount of white blood cells increases rapidly. Symptoms of AMML often include fatigue (due to anemia) or easy bruising or bleeding (due to thrombocytopenia). The cause of AMML is currently unknown. Treatment typically consists of chemotherapy.
FKBP14 kyphoscoliotic Ehlers-Danlos syndrome ( FKBP14 -kEDS) is characterized by congenital muscle hypotonia and weakness (typically improving during childhood), progressive scoliosis, joint hypermobility, hyperelastic skin, gross motor developmental delay, myopathy, and hearing impairment. ... Diagnosis Formal clinical diagnostic criteria for FKBP14 Kyphoscoliotic Ehlers-Danlos syndrome ( FKBP14 -kEDS) were established in the 2017 revised Ehlers-Danlos syndrome (EDS) nosology [Malfait et al 2017]; see Establishing the Diagnosis. ... Molecular Genetic Testing Used in FKBP14 Kyphoscoliotic Ehlers-Danlos Syndrome View in own window Gene 1 Method Proportion of Pathogenic Variants 2 Detectable by Method FKBP14 Sequence analysis 3 24/24 4, 5 Gene-targeted deletion/duplication analysis 6 Unknown 7 1. ... Nomenclature FKBP14- kEDS was initially referred to as a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. ... Management Evaluations Following Initial Diagnosis To establish the extent of disease and needs in an individual diagnosed with FKBP14 kyphoscoliotic Ehlers-Danlos syndrome ( FKBP14- kEDS), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
These include: Some heart and migraine medicines Hormone medicines, such as estrogen Antibiotics Pseudoephedrine Opioids Certain medicines for irritable bowel syndrome Chemotherapy medicines Risk factors Risk factors for ischemic colitis include: Age.
PMID 16570735 . ^ Sinding-Larsen and Johansson syndrome at Who Named It? ^ Lucena G. L., Gomes C. ... "Prevalence and Associated Factors of Osgood-Schlatter Syndrome in a Population-Based Sample of Brazilian Adolescents". ... PMID 3508010 . ^ a b Gholve PA, Scher DM, Khakharia S, Widmann RF, Green DW (2007). "Osgood Schlatter syndrome". Curr. Opin. Pediatr . 19 (1): 44–50. doi : 10.1097/MOP.0b013e328013dbea . ... "Prevalence and Associated Factors of Osgood-Schlatter Syndrome in a Population-Based Sample of Brazilian Adolescents" . ... "Prevalence and Associated Factors of Osgood-Schlatter Syndrome in a Population-Based Sample of Brazilian Adolescents" .
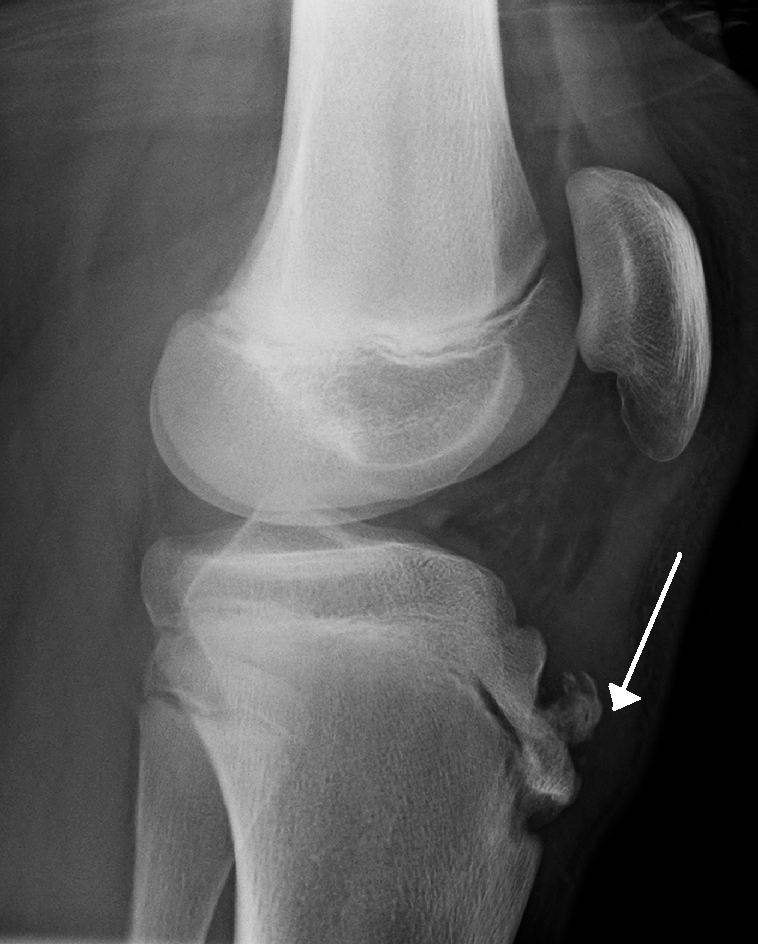
Osgood-Schlatter disease is a traction apophysitis of the anterior tibial tubercle described in active adolescents and characterized by gradual onset of pain and swelling of the anterior knee causing limping that usually disappears at the end of growth.
Please help improve it to make it understandable to non-experts , without removing the technical details. ( June 2020 ) ( Learn how and when to remove this template message ) total color blindness Achromatopsia Known as Total color blindness Specialty Ophthalmology Symptoms Day blindness , involuntary eye movement , lazy eye , photophobia Causes Acquired malfunction of the retinal phototransduction pathway Congenital damage to the diencephalon , thalamus , or the cerebral cortex Diagnosis Electroretinography Frequency 1 / 30,000 × 100% = 0.00333333333333% Achromatopsia , also known as total color blindness , is a medical syndrome that exhibits symptoms relating to at least five conditions. The term may refer to acquired conditions such as cerebral achromatopsia , but it typically refers to an autosomal recessive congenital color vision condition , the inability to perceive color and to achieve satisfactory visual acuity at high light levels, typically exterior daylight. The syndrome is also present in an incomplete form which is more properly defined as dyschromatopsia. ... Contents 1 Signs and symptoms 1.1 Complete achromatopsia 1.2 Incomplete achromatopsia 2 Cause 2.1 Acquired 2.2 Congenital 3 Pathophysiology 3.1 ACHM2 3.2 ACHM3 3.3 ACHM4 4 Management 5 Epidemiology 6 Terminology 6.1 Related 7 References 7.1 Footnotes 7.2 Sources 8 External links Signs and symptoms [ edit ] The syndrome is frequently noticed first in children around six months of age by their photophobic activity or their nystagmus . The nystagmus becomes less noticeable with age but the other symptoms of the syndrome become more relevant as school age approaches. ... The congenital forms of the condition are considered stationary and do not worsen with age. [ citation needed ] The five symptoms associated with achromatopsia or dyschromatopsia are: [ citation needed ] Achromatopsia Amblyopia – reduced visual acuity Hemeralopia – with the subject exhibiting photophobia Nystagmus Iris operating abnormalities The syndrome of achromatopsia or dyschromatopsia is poorly described in current medical and neuro-ophthalmological texts.
A number sign (#) is used with this entry because of evidence that the form of recessive achromatopsia present in high incidence among Pingelapese islanders, here designated achromatopsia-3 (ACHM3), is caused by homozygous or compound heterozygous mutation in the CNGB3 gene (605080), which encodes the beta subunit of the cone cyclic nucleotide-gated cation channel, on chromosome 8q21. A form of achromatopsia designated ACHM1 was later found to be the same as ACHM3, caused by mutation in the CNGB3 gene (605080.0002). For a general description and a discussion of genetic heterogeneity of achromatopsia, see ACHM2 (216900). Clinical Features Brody et al. (1970) described in Pingelapese people of the eastern Caroline Islands in the Pacific, a severe ocular abnormality manifested by horizontal pendular nystagmus, photophobia, amaurosis, colorblindness, and gradually developing cataract. From 4 to 10% of Pingelapese people are blind from infancy. Segregation analysis and equal sex distribution supported recessive inheritance.
A number sign (#) is used with this entry because of evidence that retinal cone dystrophy with supernormal rod electroretinogram (RCD3A) can be caused by mutation in the gene encoding the gamma subunit of cone cGMP-phosphodiesterase (PDE6H; 601190) on chromosome 12p13. In addition, achromatopsia-6 (ACHM6) can be caused by homozygous mutation in PDE6H. Another form of cone dystrophy with supernormal rod electroretinogram (RCD3B; 610356) is caused by mutation in the KCNV2 gene (607604). Clinical Features Cone Dystrophy With Supernormal Rod Electroretinogram Cone dystrophy with supernormal rod electroretinogram, also known as retinal cone dystrophy-3 (RCD3), is an autosomal recessive disorder that causes lifelong visual loss combined with a supernormal ERG response to a bright flash of light. The disorder was first described by Gouras et al. (1983) and is characterized by reduced visual acuity, photoaversion, night blindness, and abnormal color vision.
A number sign (#) is used with this entry because achromatopsia-4 can be caused by homozygous or compound heterozygous mutation in the GNAT2 gene (139340) on chromosome 1p13. Description Achromatopsia, also referred to as rod monochromacy, is an autosomal recessive ocular disorder characterized by total colorblindness, low visual acuity, photophobia, and nystagmus (Kohl et al., 2002). For a general description and a discussion of genetic heterogeneity of achromatopsia, see 216900. Inheritance Achromatopsia-4 is an autosomal recessive disorder (Kohl et al., 2002). Molecular Genetics Kohl et al. (2002) reported 5 families with achromatopsia, with 4 showing homozygosity for protein-truncation mutations in the GNAT2 gene (139340.0001; 139340.0003-139340.0004).
In very young individuals Bradyopsia; delayed cone adaptation RGS9 Prolonged electroretinal response suppression leading to difficulties adjusting to changes in luminance Normal to subnormal visual acuity Photophobia Alström syndrome 11 ALMS1 AR Infantile nystagmus Photophobia Severely reduced visual acuity Poor or no color discrimination Possible additional findings in Alström syndrome: cardiomyopathy, kidney failure, obesity, sensorineural hearing loss, diabetes In young individuals AD = autosomal dominant; AR = autosomal recessive; ERG = electroretinogram; MOI = mode of inheritance; XL = X-linked 1.
A number sign (#) is used with this entry because of evidence that achromatopsia-7 (ACHM7) is caused by homozygous or compound heterozygous mutation in the ATF6 gene (605537) on chromosome 1q23. Description Achromatopsia (ACHM) is an autosomal recessive disorder resulting from lack of cone photoreceptor function. Affected individuals present from birth or early infancy with photophobia, nystagmus, severely reduced visual acuity, and color blindness (summary by Kohl et al., 2015). For a general description and a discussion of genetic heterogeneity of achromatopsia, see ACHM2 (216900). Clinical Features Ansar et al. (2015) studied 2 sibs, their female cousin (the proband), and their nephew from a multiply consanguineous Pakistani family.
A rare autosomal recessive retinal disorder characterized by color blindness, nystagmus, photophobia, and severely reduced visual acuity due to the absence or impairment of cone function. Epidemiology The prevalence is estimated to be 1/30,000-1/50,000 worldwide. Clinical description ACHM is characterized by reduced visual acuity, pendular nystagmus, increased sensitivity to light (photophobia), a small central scotoma, and reduced or complete loss of color discrimination. Most individuals have complete ACHM, with total lack of function in all three types of cones. Rarely, individuals have incomplete ACHM, with similar, but generally less severe symptoms.
A number sign (#) is used with this entry because of evidence that complete achromatopsia and some cases of incomplete achromatopsia are caused by homozygous or compound heterozygous mutation in the CNGA3 gene (600053), which encodes the alpha subunit of the cone photoreceptor cGMP-gated cation channel, on chromosome 2q11. Description Total colorblindness, also referred to as rod monochromacy or complete achromatopsia, is a rare congenital autosomal recessive disorder characterized by photophobia, reduced visual acuity, nystagmus, and the complete inability to discriminate between colors. Electroretinographic recordings show that in achromatopsia the rod photoreceptor function is normal, whereas cone photoreceptor responses are absent (summary by Kohl et al., 1998). Genetic Heterogeneity of Total Achromatopsia A form of achromatopsia previously designated achromatopsia-1 (ACHM1) was later found to be the same as achromatopsia-3 (ACHM3; 262300), caused by mutation in the CNGB3 gene (605080). ACHM4 (613856) is caused by mutation in the GNAT2 gene (139340); ACHM5 (613093) is caused by mutation in the PDE6C gene (600827); ACHM6 (see 610024) is caused by mutation in the PDE6H gene (601190); and ACHM7 (616517) is caused by mutation in the ATF6 gene (605537).
Maumenee (1982) questioned the existence of mendelian distichiasis except as part of the syndrome of lymphedema with distichiasis (153400). ... A similar but distinct eyelash anomaly was reported in association with the Setleis forceps marks syndrome (227260) by Frederick and Robb (1992).
Isolated distichiasis is a rare congenital eyelid anomaly characterized by an accessory row of eyelashes (that may be partial or complete) posterior to the normal row of cilia, at or close to the meibomian gland orifices, that is not associated with any other condition, and that may lead to ocular irritation and corneal damage if left untreated.
Lolin et al. (1989) described a 7-year-old girl who was found to have cryofibrinogenemia after developing transient nephrotic syndrome and hematuria after anesthesia. ... The mutation in this disorder may involve one of the structural genes for fibrinogen (134820, 134830, 134850). GU - Transient nephrotic syndrome - Hematuria Inheritance - Autosomal dominant Lab - Cryofibrinogenemia Skin - Acrocyanosis with cold exposure ▲ Close
Alternatives are that it is a pleiotropic manifestation of the gene responsible for either infantile neuroaxonal dystrophy or infantile osteopetrosis or that it represents a contiguous gene syndrome combining features of the 2 recessive disorders. ... Autosomal recessive vs contiguous gene syndrome ▲ Close
A number sign (#) is used with this entry because of evidence that autosomal recessive osteopetrosis-5 (OPTB5) is caused by homozygous mutation in the gene encoding osteopetrosis-associated transmembrane protein-1 (OSTM1; 607649) on chromosome 6q21. For a general phenotypic description and a discussion of genetic heterogeneity of autosomal recessive osteopetrosis, see OPTB1 (259700). Description Autosomal recessive osteopetrosis-5 is a form of infantile malignant osteopetrosis, characterized by defective osteoclast function resulting in decreased bone resorption and generalized osteosclerosis. Defective resorption causes development of densely sclerotic fragile bones and progressive obliteration of the marrow spaces and cranial foramina. Marrow obliteration is associated with extramedullary hematopoiesis and hepatosplenomegaly, and results in anemia and thrombocytopenia, whereas nerve entrapment accounts for progressive blindness and hearing loss.
Osteopetrosis is a bone disease that makes bones abnormally dense and prone to breakage (fracture). Researchers have described several major types of osteopetrosis, which are usually distinguished by their pattern of inheritance: autosomal dominant, autosomal recessive, or X-linked. The different types of the disorder can also be distinguished by the severity of their signs and symptoms. Autosomal dominant osteopetrosis (ADO), which is also called Albers-Schönberg disease, is typically the mildest type of the disorder. Some affected individuals have no symptoms. In these people, the unusually dense bones may be discovered by accident when an x-ray is done for another reason.
Although the disorder had features resembling Stickler syndrome (108300), SED congenita (183900), and some other disorders, MacDermot et al. (1987) thought it was distinct. It may, however, be allelic to either Stickler syndrome or SED congenita. The authors believed it differed from Namaqualand hip dysplasia (604864) because deafness and eye abnormalities are not features of the latter condition.
Spondyloepiphyseal dysplasia (SED), MacDermot type is characterized by short stature, femoral epiphyseal dysplasia, mild vertebral changes and sensorineural deafness. Epidemiology The syndrome has been described in a family in which females in four successive generations were affected.