In the first family, an affected mother and son both showed left ventricular hypertrophy (LVH) on electrocardiography (ECG), and the 62-year-old mother also exhibited Wolff-Parkinson-White syndrome (see 194200) whereas her 29-year-old son had abnormal Q-waves. ... INHERITANCE - Autosomal dominant CARDIOVASCULAR Heart - Left ventricular hypertrophy (in some patients) - Sigmoid septal shape (in some patients) - Abnormal Q-waves on electrocardiography (in some patients) - Wolff-Parkinson-White syndrome (rare) - Sudden cardiac death after exercise (in some patients) - Myocyte hypertrophy - Interstitial fibrosis MOLECULAR BASIS - Caused by mutation in the titin-CAP gene (TCAP, 604488.0003 ) ▲ Close
Immunotactoid glomerulopathy (ITG) is a very rare condition characterized by glomerular accumulation of microtubules in the mesangium and the glomerular basement membrane, that mainly presents with proteinuria, micro-hematuria, nephrotic syndrome, renal insufficiency and hematologic malignancy. ... Epidemiology Immunotactoid glomerulopathy (ITG) is a very rare condition characterized by glomerular accumulation of microtubules in the mesangium and the glomerular basement membrane, that mainly presents with proteinuria, micro-hematuria, nephrotic syndrome, renal insufficiency and hematologic malignancy.
Immunotactoid glomerulopathy , also known as glomerulonephritis with organized monoclonal microtubular immunoglobulin deposits (GOMMID), is a very uncommon cause of glomerular disease . It is related to a similar disease known as fibrillary glomerulopathy , which is more common. Both disorders probably result from deposits derived from immunoglobulins, but in most cases the cause is idiopathic (unknown). On electron microscopy , immunotactoid glomerulopathy is characterized by the formation of microtubules which are much larger than the fibrils observed in fibrillary glomerulonephritis (30 to 50 versus 16 to 24 nm in diameter). The signs and symptoms include blood (hematuria) and protein (proteinuria) in the urine, kidney insufficiency and high blood pressure.
Edemas may involve the digestive tract resulting in a clinical picture similar to that seen in intestinal occlusion syndrome, sometimes associated with ascites and hypovolemic shock. ... Differential diagnosis The differential diagnosis should include acquired angioedema (see this term), intestinal occlusion syndrome and histamine-induced angioedema (of allergenic or nonallergenic origin) generally associated with urticaria.
A number sign (#) is used with this entry because of evidence that hereditary angioedema type III (HAE3) is caused by heterozygous mutation in the gene encoding coagulation factor XII (F12; 610619) on chromosome 5q35. Description Hereditary angioedema type III is a rare disorder characterized clinically by recurrent skin swelling, abdominal pain attacks, and potentially life-threatening upper airway obstruction. HAE III occurs almost exclusively in women and is often precipitated or worsened by high estrogen levels (e.g., during pregnancy or treatment with oral contraceptives). It differs from HAE types I and II (106100) in that both concentration and function of C1 inhibitor (C1NH; 606860) are normal (summary by Dewald and Bork, 2006). Clinical Features Binkley and Davis (2000) reported a 3-generation Italian family with a unique type of hereditary angioedema that was estrogen-dependent.
Hereditary angioedema (HAE) is a disease characterized by recurrent episodes (also called attacks) of severe swelling of the skin and mucous membranes. The age at which attacks begin varies, but most people have their first one in childhood or adolescence. The frequency of attacks usually increases after puberty. Attacks most often affect 3 parts of the body: Skin - the most common sites are the face (such as the lips and eyes), hands, arms, legs, genitals, and buttocks. Skin swelling can cause pain, dysfunction, and disfigurement, although it is generally not dangerous and is temporary. Gastrointestinal tract - the stomach, intestines, bladder, and/or urethra may be involved.
Diagnosis [ edit ] Differential diagnosis [ edit ] Common variable immunodeficiency IPEX syndrome Management [ edit ] Along with treatment for infections and other complications several additional treatments have been tried. ... Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity.
O'Driscoll et al. (2010) noted some phenotypic overlap with Hennekam syndrome (235510) and FG syndrome (305450), but concluded that the patients did not fit into either category, suggesting a new distinct disorder.
Differential diagnosis Differential diagnoses include renal PHA1, congenital adrenal hyperplasia (CAH) associated with 21-hydroxylase deficiency or 3-beta-hydroxysteroid dehydrogenase deficiency, hypoaldosteronism (HA) due to aldosterone deficiency, antenatal or infantile Bartter syndrome (in particular, type 2 Bartter syndrome caused by ROMK mutations) and transient PHA (see these terms).
A milder, autosomal dominant form of type I pseudohypoaldosteronism (PHA1A; 177735) is caused by mutations in the mineralocorticoid receptor gene (MCR, NR3C2; 600983). Gitelman syndrome (263800), another example of primary renal tubular salt wasting, is due to mutation in the thiazide-sensitive sodium-chloride cotransporter (SLC12A3; 600968). ... Chang et al. (1996) studied these genes because mutations resulting in constitutive activation of ENaC activity were demonstrated to cause Liddle syndrome (177200), an autosomal dominant form of hypertension characterized by volume expansion, hypokalemia, and alkalosis.
Autosomal recessive pseudohypoaldosteronism type 1 is a disorder of electrolyte metabolism characterized by excess loss of salt in the urine and high concentrations of sodium in sweat, stool, and saliva. The disorder involves multiple organ systems and is especially dangerous in the newborn period. Laboratory tests may show hyponatremia , hyperkalemia , and increased plasma renin activity with high levels of aldosterone in the blood. Respiratory tract infections are common in affected children. Treatment involves aggressive salt replacement and control of hyperkalemia. The disorder may become less severe with age. Autosomal recessive pseudohypoaldosteronism type 1 (PHA1B) is transmitted in an autosomal recessive manner and is caused by mutations in the genes coding for the subunits of the amiloride-sensitive sodium channel ( SCNN1A , SCNN1B and SCNN1G ).
'Autism spectrum disorder,' sometimes referred to as ASD, is a broader phenotype encompassing the less severe disorders Asperger syndrome (see ASPG1; 608638) and pervasive developmental disorder, not otherwise specified (PDD-NOS). ... Mental retardation coexists in approximately two-thirds of individuals with ASD, except for Asperger syndrome, in which mental retardation is conspicuously absent (Jones et al., 2008).
A rare acute myeloid leukemia characterized by primary differentiation to basophils. Microscopically, peripheral blood and bone marrow blasts contain coarse cytoplasmic basophilic granules which are positive with metachromatic staining (toluidine blue). Electron microscopy confirms that granules show features characteristic of basophil precursors. Mature basophils are usually sparse. Patients may present with manifestations related to bone marrow failure, as well as hepatosplenomegaly, cutaneous involvement, lytic lesions, and hyperhistaminemia. The disease is associated with a poor prognosis.
According to this, it can be divided into Anisocytosis with microcytosis – Iron deficiency , sickle cell anemia Anisocytosis with macrocytosis – Folate or vitamin B 12 deficiency , autoimmune hemolytic anemia , cytotoxic chemotherapy, chronic liver disease , myelodysplastic syndrome Increased RDW is seen in iron deficiency anemia and decreased or normal in thalassemia major (Cooley's anemia), thalassemia intermedia Anisocytosis with normal RBC size – Early iron, vit B12 or folate deficiency, dimorphic anemia , Sickle cell disease, chronic liver disease, myelodysplastic syndrome [2] Etymology [ edit ] From Ancient Greek : an - without, or negative quality, iso - equal, cyt - cell, - osis condition. [3] See also [ edit ] Anisopoikilocytosis Poikilocytosis References [ edit ] ^ Barbara J.
HMG-CoA lyase deficiency is sometimes mistaken for Reye syndrome, a severe disorder that develops in children while they appear to be recovering from viral infections such as chicken pox or flu. Most cases of Reye syndrome are associated with the use of aspirin during these viral infections.
Differential diagnosis Differential diagnosis includes sepsis, fatty acid oxidation disorders, organic acidurias and Reye's syndrome. Antenatal diagnosis During the third trimester of gestation, amniotic fluid organic acid levels, as well as maternal urinalysis may indicate 3HMG; confirmation requires testing of cultured amniocytes or chorionic villi for molecular study.
Act Early.” campaign - Information for parents on early childhood development and developmental disabilities "Recognizing Developmental Delays in Children" , WebMD , retrieved 31 May 2019 v t e Malnutrition Protein-energy malnutrition Kwashiorkor Marasmus Catabolysis Vitamin deficiency B vitamins B 1 Beriberi Wernicke–Korsakoff syndrome Wernicke's encephalopathy Korsakoff's syndrome B 2 Riboflavin deficiency B 3 Pellagra B 6 Pyridoxine deficiency B 7 Biotin deficiency B 9 Folate deficiency B 12 Vitamin B 12 deficiency Other A: Vitamin A deficiency Bitot's spots C: Scurvy D: Vitamin D deficiency Rickets Osteomalacia Harrison's groove E: Vitamin E deficiency K: Vitamin K deficiency Mineral deficiency Sodium Potassium Magnesium Calcium Iron Zinc Manganese Copper Iodine Chromium Molybdenum Selenium Keshan disease Growth Delayed milestone Failure to thrive Short stature Idiopathic General Anorexia Weight loss Cachexia Underweight This medical sign article is a stub .
In most cases, spina bifida occulta causes no symptoms and doesn't need treatment. Tethered cord syndrome. The spinal cord normally hangs freely within the spinal canal. Tethered cord syndrome is a disorder that occurs when tissue attached to the spinal cord limits its movements.
Small depression in the skin located just above the buttocks This article needs more medical references for verification or relies too heavily on primary sources . Please review the contents of the article and add the appropriate references if you can. Unsourced or poorly sourced material may be challenged and removed . Find sources: "Sacral dimple" – news · newspapers · books · scholar · JSTOR ( February 2014 ) A sacral dimple (also termed pilonidal dimple or spinal dimple ) [1] is a small depression in the skin, located just above the buttocks . [2] [3] [4] [5] The name comes from the sacrum , the bone at the end of the spine, over which the dimples are found. Sacral dimples are rare, occurring in up to 4% of the population. [1] [5] The majority of these dimples are minor and do not represent any underlying disease; [1] [3] [5] however, the minority may be a sign of disease, notably spina bifida . [3] [5] Even so, this is usually the spina bifida occulta form, which is the least serious kind. [3] Additionally, a sacral dimple may be indicative of a possible kidney problem that can be checked with an ultrasound . Sacral dimples are usually spotted in post-natal checks by a pediatrician , [3] [5] who will check: whether the floor of the dimple can be seen to be covered with skin; whether there is a tuft of hair in the dimple; whether there are any other problems such as weak lower limbs; the distance from the buttocks to the dimple (closer is better).
Tlili et al. (2005) reported a consanguineous Tunisian family in which 4 sibs had congenital profound hearing loss (greater than 90 dB) but were otherwise healthy with no dysmorphic or other abnormal findings indicative of syndromic deafness. No vestibular defects were detected. ... The whirler mutation in the mouse causes, in the homozygous adult, the shaker-waltzer syndrome: deafness and circling with tossing of the head (Fleming et al., 1994).
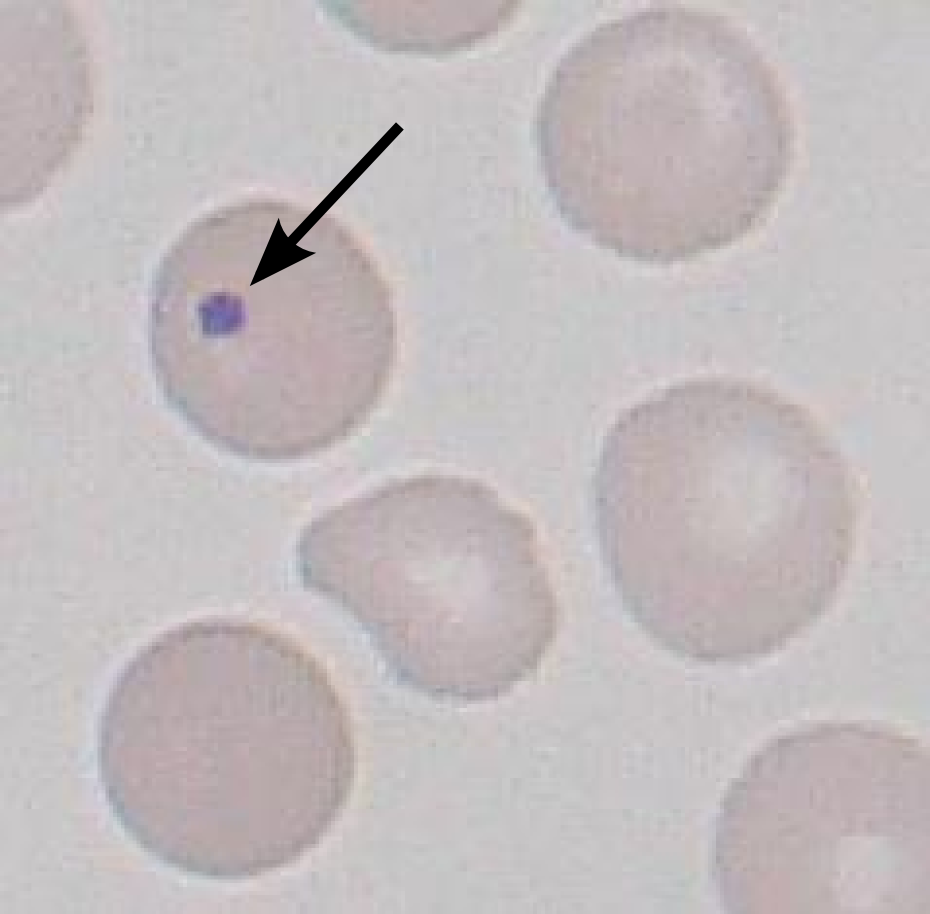
Common causes include asplenia (post-splenectomy) or congenital absence of spleen ( heterotaxy syndrome with asplenia ). Spleens are also removed for therapeutic purposes in conditions like hereditary spherocytosis , trauma to the spleen, and autosplenectomy caused by sickle cell anemia . ... Howell–Jolly bodies are also seen in amyloidosis , severe hemolytic anemia , megaloblastic anemia , hereditary spherocytosis, and myelodysplastic syndrome (MDS). The bodies can also can be seen in premature infants.
She never had loss of consciousness. Her epilepsy syndrome was classified as juvenile myoclonic epilepsy. ... Escayg et al. (2000) also reported a German father and son with an atypical but similar clinical syndrome of idiopathic generalized epilepsy with rare juvenile atypical prolonged absences and occasional generalized tonic-clonic seizures (GTCS).
A number sign (#) is used with this entry because of evidence that susceptibility to idiopathic generalized epilepsy-13 (EIG13), including juvenile myoclonic epilepsy-5 (EJM5) and childhood absence epilepsy-4 (ECA4), can be conferred by heterozygous mutation in the GABRA1 gene (137160) on chromosome 5q34. Description Childhood absence epilepsy and juvenile myoclonic epilepsy are both subtypes of what has classically been called idiopathic generalized epilepsy (IGE, EIG; see 600669). For a phenotypic description and a discussion of genetic heterogeneity of idiopathic generalized epilepsy, see 600669. For a phenotypic description and a discussion of genetic heterogeneity of juvenile myoclonic epilepsy and childhood absence epilepsy, see ECA1 (600131) and JME (254770), respectively. Clinical Features Cossette et al. (2002) reported a French Canadian family in which 14 members over 4 generations had juvenile myoclonic epilepsy.
For a phenotypic description and a discussion of genetic heterogeneity of idiopathic generalized epilepsy (IGE), see 600669. Juvenile myoclonic epilepsy (JME; see 254770) is a form of idiopathic generalized epilepsy. Mapping Elmslie et al. (1997) tested for linkage between JME and chromosomal regions harboring genes for nAChR subunits in 34 pedigrees using parametric and nonparametric analyses. Strong evidence for linkage with heterogeneity was found to polymorphic loci encompassing the region containing the gene that encodes the alpha-7 subunit of nAChR (CHRNA7; 118511), which maps to chromosome 15q14. Elmslie et al. (1997) suggested that this major locus (EJM2) contributes to genetic susceptibility to JME in a majority of the families studied.
Juvenile myoclonic epilepsy is the most common hereditary idiopathic generalized epilepsy syndrome and is characterized by myoclonic jerks of the upper limbs on awakening, generalized tonic-clonic seizures manifesting during adolescence and triggered by sleep deprivation, alcohol intake, and cognitive activities, and typical absence seizures (30% of cases).
For a phenotypic description and a discussion of genetic heterogeneity of juvenile myoclonic epilepsy (JME), see 254770. JME is a form of idiopathic generalized epilepsy (IGE; 600669). Clinical Features Kapoor et al. (2007) reported a family from southern India in which 8 individuals had juvenile myoclonic epilepsy inherited in an autosomal dominant pattern. The proband was a 32-year-old woman who developed morning myoclonic jerks at age 14 years and generalized tonic-clonic seizures at age 20. EEG recordings showed polyspike and wave discharges characteristic of a generalized epilepsy. Other affected members had a similar history. None had absence or febrile seizures.
Juvenile myoclonic epilepsy Other names Janz syndrome Specialty Neurology Juvenile myoclonic epilepsy (JME), also known as Janz syndrome , is a fairly common form of generalized epilepsy of presumed genetic origin (previously known an idiopathic generalized epilepsy ), [1] representing 5-10% of all epilepsy cases. [2] [3] [4] This disorder typically first manifests itself between the ages of 12 and 18 with sudden brief involuntary single or multiple episodes of muscle(s) contractions caused by an abnormal excessive or synchronous neuronal activity in the brain. ... Sleep deprivation is a major factor in triggering seizures in JME patients. [5] Cause [ edit ] Juvenile myoclonic epilepsy is an inherited genetic syndrome, but the way in which this disorder is inherited is unclear. ... The primary diagnosis for JME is a good knowledge of patient history and the neurologist's familiarity with the myoclonic jerks, which are the hallmark of the syndrome. [16] Additionally, an electroencephalogram (EEG) , will indicate a characteristic pattern of waves and spikes associated with the syndrome such as generalized 4–6 Hz polyspike and slow wave discharges.
Juvenile myoclonic epilepsy is an epilepsy syndrome characterized by myoclonic jerks (quick jerks of the arms or legs), generalized tonic-clonic seizures (GTCSs), and sometimes, absence seizures .
A number sign (#) is used with this entry because of evidence that susceptibility to juvenile myoclonic epilepsy-10 (EJM10) is conferred by heterozygous mutation in the ICK gene (612325) on chromosome 6p12. Description Juvenile myoclonic epilepsy-10 is an autosomal dominant seizure disorder with variable manifestations, even within families. Affected individuals have febrile, myoclonic, tonic-clonic, or absence seizures, although several seizure types can occur in the same individual. The age of onset also shows great variability: some patients present in the first years of life, whereas other have onset of seizures in teenage years. EEG typically shows 3.5 to 5 Hz polyspike wave discharges. There is evidence of incomplete penetrance (summary by Bailey et al., 2018).
For general phenotypic information and a discussion of genetic heterogeneity of juvenile myoclonic epilepsy, see 254770. Clinical Features Ratnapriya et al. (2010) reported a 4-generation family from southern India in which 6 living members had juvenile myoclonic epilepsy. Age at onset ranged from 12 to 20 years, and all had myoclonic seizures with secondary generalized tonic-clonic seizures. Two patients had a history of febrile seizures, and 2 had absence seizures. EEG showed generalized 4-6 Hz polyspike and wave discharges characteristic of a generalized epilepsy.
A number sign (#) is used with this entry because some evidence has suggested that susceptibility to idiopathic generalized epilepsy-11 (EIG11), juvenile myoclonic epilepsy-8 (EJM8), and juvenile absence epilepsy-2 (EJA2) may be conferred by variation in the chloride channel-2 gene (CLCN2; 600570) on chromosome 3q27. However, there has been some controversy over whether variation in the CLCN2 gene has a role in epilepsy (see MOLECULAR GENETICS). Description Both juvenile myoclonic epilepsy and juvenile absence epilepsy are subtypes of idiopathic generalized epilepsy (EIG). For a general phenotypic description and a discussion of genetic heterogeneity of these disorders, see EIG (600669), EJM (254770), and EJA (607631). Mapping Sander et al. (2000) used nonparametric multipoint linkage analysis to identify susceptibility loci among 130 IGE-multiplex families ascertained through a proband with childhood or juvenile absence epilepsy or juvenile myoclonic epilepsy, and 1 or more sibs affected by an IGE trait.
For general phenotypic information and a discussion of genetic heterogeneity of juvenile myoclonic epilepsy, see 254770. Mapping Greenberg et al. (1987) studied 24 families with JME. Segregation analysis allowed them to reject fully penetrant dominant and recessive models. For the linkage analysis, they assumed a fully penetrant recessive model or a recessive model with 60% penetrance. Using either clinical phenotype or EEG changes to score persons as affected with JME, Greenberg et al. (1987, 1988) found evidence for linkage to BF (138470) and HLA (142800) on chromosome 6p21; the lod score exceeded 3.0 when HLA and BF were used together. Greenberg et al. (1989) and Delgado-Escueta et al. (1989) presented additional evidence for linkage to HLA and BF.
Winawer et al. (2003) concluded that there are distinct genetic effects on absence and myoclonic seizures, and suggested that examining seizure types as opposed to syndromes may be more useful in linkage studies.
A number sign (#) is used with this entry because of evidence that susceptibility to idiopathic generalized epilepsy-10 (EIG10), generalized epilepsy with febrile seizures plus, type 5 (GEFSP5), and juvenile myoclonic epilepsy-7 (EJM7) can be conferred by variation in the GABRD gene (137163) on chromosome 1p36.3. Description Idiopathic generalized epilepsy (EIG) is a broad term that encompasses several common seizure phenotypes, classically including childhood absence epilepsy (CAE, ECA), juvenile absence epilepsy (JAE), and juvenile myoclonic epilepsy (JME, EJM) (Commission on Classification and Terminology of the International League Against Epilepsy, 1989). Generalized epilepsy with febrile seizures plus (GEFS+) shows phenotypic overlap with IGE, and includes patients with early-onset febrile seizures who later develop various types of febrile and afebrile seizures, such as those observed in EIG (summary by Singh et al., 1999). For a general phenotypic description and a discussion of genetic heterogeneity of EIG, see 600669. For a general phenotypic description and a discussion of genetic heterogeneity of GEFS+, see 604233.
This disorder is sometimes mistaken for Reye syndrome, a severe disorder that may develop in children while they appear to be recovering from viral infections such as chicken pox or flu. Most cases of Reye syndrome are associated with the use of aspirin during these viral infections.
Beginning at 1 year of age, she had suffered 3 severe Reye syndrome-like episodes precipitated by mild viral illnesses. ... The patients were first seen between 8 and 18 months of age with recurrent episodes of hypoketotic hypoglycemia accompanied by a decreased level of consciousness and hepatomegaly. One patient had 2 Reye syndrome-like episodes. The patients were successfully treated with medium-chain triglycerides and avoidance of fasting. ... INHERITANCE - Autosomal recessive CARDIOVASCULAR Heart - Cardiomegaly - Cardiac rhythm disturbances ABDOMEN Liver - Hepatomegaly Gastrointestinal - Poor feeding - Diarrhea MUSCLE, SOFT TISSUES - Muscle weakness is not a feature NEUROLOGIC Central Nervous System - Hypotonia - Lethargy - Seizures - Coma - Encephalopathy, recurrent METABOLIC FEATURES - Hypoketotic hypoglycemia - Renal tubular acidosis PRENATAL MANIFESTATIONS Maternal - Acute fatty liver in pregnancy (fetus with carnitine palmitoyltransferase I (CPT1) deficiency) - HELLP syndrome LABORATORY ABNORMALITIES - Mild to moderate hyperammonemia - Transient hyperlipidemia - Elevated creatine kinase - Elevated transaminases - No dicarboxylic aciduria - No ketonuria - Normal to elevated total plasma carnitine - Elevated free carnitine - Carnitine palmitoyltransferase I deficiency (fibroblast, liver, leukocytes) - Decreased CPT1 activity - Decreased long-chain fatty acid oxidation MISCELLANEOUS - Onset
Postnatal clinical findings Hepatic encephalopathy (similar to that seen in Reye syndrome) precipitated by fasting or fever (see Supportive laboratory findings ) Rapid onset of symptoms in association with a relatively common infectious disease, such as a febrile or gastrointestinal illness Supportive laboratory findings Hypoketotic hypoglycemia , defined as low blood glucose concentration (<40 mg/dL) in the absence of ketone bodies in the urine Elevated liver enzymes.
Differential diagnosis Differential diagnosis includes fatty acid and ketogenesis disorders such as medium-chain acyl-CoA dehydrogenase (MCAD deficiency; see this term), other long-chain fatty acid oxidation disorders such as carnitine palmitoyltransferase (CPT) 2 deficiency and Reye's syndrome (see these terms). Antenatal diagnosis Antenatal diagnosis is possible by mutational analysis if the mutations in a proband have been identified.
Differential diagnosis [ edit ] This condition is sometimes mistaken for fatty acid and ketogenesis disorders such as Medium-chain acyl-coenzyme A dehydrogenase deficiency (MCAD), other long-chain fatty acid oxidation disorders such as Carnitine palmitoyltransferase II deficiency (CPT-II) and Reye syndrome . [6] Treatment [ edit ] Treatment for this condition is supportive. ... National Library of Medicine v t e Inborn error of lipid metabolism : fatty-acid metabolism disorders Synthesis Biotinidase deficiency (BTD) Degradation Acyl transport Carnitine CPT1 CPT2 CDSP CACTD Adrenoleukodystrophy (ALD) Beta oxidation General Acyl CoA dehydrogenase Short-chain SCADD Medium-chain MCADD Long-chain 3-hydroxy LCHAD Very long-chain VLCADD Mitochondrial trifunctional protein deficiency (MTPD): Acute fatty liver of pregnancy Unsaturated 2,4 Dienoyl-CoA reductase deficiency (DECRD) Odd chain Propionic acidemia (PCC deficiency) Other 3-hydroxyacyl-coenzyme A dehydrogenase deficiency (HADHD) Glutaric acidemia type 2 (MADD) To acetyl-CoA Malonic aciduria (MCD) Aldehyde Sjögren–Larsson syndrome (SLS) v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
Carnitine palmitoyltransferase I deficiency (CPT1A deficiency) is an inherited metabolic condition that prevents the body from converting certain fats (long-chain fatty acids) into energy, particularly during periods without food. Carnitine, a natural substance acquired mostly through the diet, is required by cells to process fats and produce energy. Symptoms of this condition often appear early in life and include low blood sugar (hypoglycemia) and low levels of ketones, which are produced when the body breaks down fat for energy (hypoketotic hypoglycemia). This can lead to a greater risk for loss of consciousness or seizures. People with this disorder typically also have an enlarged liver (hepatomegaly), muscle weakness, nervous system damage, and elevated levels of carnitine in the blood.