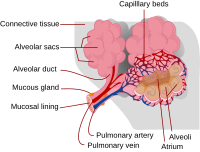
Causes of acute alveolar lung disease include pulmonary edema (cardiogenic or neurogenic), pneumonia (bacterial or viral), systemic lupus erythematosus , bleeding in the lungs (e.g., Goodpasture syndrome ), idiopathic pulmonary hemosiderosis , and granulomatosis with polyangiitis . ... Ventilatory support is recognized as an essential component to treat pulmonary edema and acute respiratory distress syndrome . [3] Non-invasive ventilation is the first step for patient's who require ventilatory support. ... ISBN 978-3642037085 . ^ Reyes, Felix M.; Le, Jacqueline K. (2020), "Lung Exam" , StatPearls , StatPearls Publishing, PMID 29083650 , retrieved 2020-04-19 ^ Fan, Eddy; Brodie, Daniel; Slutsky, Arthur S. (2018-02-20). "Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment" .
Suggestive Findings Mitochondrial short-chain enoyl-CoA hydratase 1 deficiency (ECHS1D) should be suspected in individuals with clinical features of Leigh syndrome and/or exercise-induced dystonia who have supportive brain MRI and biochemical findings, including early-onset lactic acidosis. ... Because the phenotype of ECHS1D is broad, individuals with the distinctive biochemical findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with lactic acidosis, Leigh syndrome, and/or dystonia are more likely to be diagnosed using genomic testing (see Option 2). ... If only one or no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications. A mitochondrial disease, Leigh syndrome, or lactic acidosis multigene panel that includes ECHS1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. ... Option 2 When the phenotype is indistinguishable from many other inherited disorders characterized by early epileptic encephalopathy, Leigh Syndrome, or lactic acidosis, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. ... Mitochondrial complex V (ATP synthase) deficiency, nuclear type 2 (OMIM 614052) TMEM70 Mitochondrial complex I deficiency due to ACAD9 deficiency (OMIM 611126) ACAD9 Other Leigh syndromes (see Nuclear Gene-Encoded Leigh Syndrome Overview & Mitochondrial DNA-Associated Leigh Syndrome and NARP) >60 genes AR mt XL T 2 hyperintensity of the basal ganglia Dystonia Developmental regression Lactic acidosis ECHS1D may be clinically indistinguishable from other Leigh syndromes.
Dangerous pseudoscientific concept This article is part of a series on Alternative medicine General information Alternative medicine Alternative veterinary medicine Quackery (Health fraud) History of alternative medicine Rise of modern medicine Pseudoscience Antiscience Skepticism Skeptical movement National Center for Complementary and Integrative Health Terminology of alternative medicine Therapeutic nihilism Fringe medicine and science Acupressure Acupuncture Alkaline diet Anthroposophic medicine Apitherapy Applied kinesiology Aromatherapy Auriculotherapy Bates method Black salve Bodywork Bonesetter Bowen technique Breathwork Fake COVID-19 treatments Cancer treatments Charcoal cleanse Chiropractic Chiropractic treatment techniques Vertebral subluxation Christian Science Chromotherapy Colon cleansing Coffee enema Colorpuncture Colloidal silver Craniosacral therapy Crystal healing Cupping therapy Dental amalgam controversy Detoxification Foot detox Ear candling Energy medicine Esoteric energy Therapeutic touch Fabunan Antiviral Injection Facilitated communication Feldenkrais Method Functional medicine Hair analysis Herbal medicine Holistic dentistry Hologram bracelet Homeopathy Bach flower remedies Biological terrain assessment Hypnotherapy Iridology Ionized jewelry Jilly Juice Lightning Process Lymphotherapy Medical intuitive Mesmerism Magnet therapy Manual therapy Megavitamin therapy Mind–body interventions MMS Myofascial release NAET Naturopathy Oil pulling Orgone Orthomolecular medicine Orthopathy Osteomyology Osteopathy Ozone therapy Parapsychology Phrenology Psychic surgery Psychodermatology Radionics Rapid prompting method RBOP Reiki Reflexology Rolfing Scientific racism ThetaHealing Thought Field Therapy Urophagia Vaginal steaming Vision therapy Vitalism Young blood transfusion Zero balancing Conspiracy theories ( list ) Big Pharma conspiracy theory HIV/AIDS denialism OPV AIDS hypothesis Anti-vaccination Vaccines and autism MMR vaccine and autism Water fluoridation controversy GMO conspiracy theories Misinformation related to the COVID-19 pandemic Classifications Alternative medical systems Mind–body intervention Biologically-based therapy Manipulative methods Energy therapy Traditional medicine African Muti Southern Africa Ayurveda Ayurvedic acupressure Dosha Maharishi Vedic Approach to Health Balneotherapy Brazilian Bush medicine Cambodian Chinese Blood stasis Chinese herbology Dit Da Gua sha Gill plate trade Meridian Moxibustion Pressure point Qi San Jiao Tui na Zang-fu Chumash Curandero Faith healing Iranian Jamu Kambo Japanese Korean Mien Shiang Mongolian Prophetic medicine Shamanism Shiatsu Siddha Sri Lankan Thai massage Tibetan Unani Vietnamese Diagnoses Adrenal fatigue Aerotoxic syndrome Candida hypersensitivity Chronic Lyme disease Electromagnetic hypersensitivity Heavy legs Leaky gut syndrome Multiple chemical sensitivity Wilson's temperature syndrome v t e " Rope worms " (or " ropeworms ") is a pseudoscientific term for long thin pieces of damaged intestinal epithelium or other bowel content that have been misidentified as human parasitic worms . [1] [2] [3] "Rope worms" were reported in 2013 in two self-published papers by Volinsky and Gubarev et al. [4] In fact, they are not actual parasites, but instead fragments of mucous membrane shed from the gut following the use of bleach enemas ( sodium chlorite mixed with citric acid, forming chlorine dioxide and marketed as Miracle Mineral Supplement ) and other similarly ineffective and toxic cleanses, such as the essential oil enema described by Volinsky et al. [3] [5] The phenomenon results from improper identification of intestinal artifacts expelled from the body. [6] These "ropeworms" are often discussed, with images shared and claimed as evidence of successful detoxing , on autism forums and altmed Facebook groups, wherein this highly toxic product (MMS) is falsely claimed to cure autism and a myriad of other conditions. [7] In one Facebook group, 8500 members have allegedly been charged $60 to join, half a million dollars combined, leading to questioning of the leaders' intentions. [8] Because they know what they are doing is not sanctioned by medical studies and is considered abusive, parents in these groups may be reluctant to take their children to their doctors, even when dangerous reactions to chlorine dioxide are apparent, such as vomiting, exhaustion, dehydration, and jaundice .
Ascites and death occurred in 4; autopsies in 2 of the deceased patients revealed hepatic vein stenosis with Budd-Chiari syndrome (see 600880). Ozen et al. (2017) studied 11 patients and 2 deceased relatives with a history of protein-losing enteropathy from 8 families of Turkish, Moroccan, or Syrian descent. ... The authors designated this disorder, comprising CD55 deficiency with hyperactivation of complement, angiopathic thrombosis, and protein-losing enteropathy, as 'CHAPLE' syndrome. Kurolap et al. (2017) reported a large consanguineous Muslim-Arab family in which 6 individuals had protein-losing enteropathy associated with hypercoagulability. ... INHERITANCE - Autosomal recessive GROWTH Other - Growth retardation HEAD & NECK Face - Edema CARDIOVASCULAR Heart - Atrial thrombosis - Ventricular thrombosis Vascular - Cerebrovascular thrombosis - Sinus vein thrombosis - Pulmonary artery thrombosis - Superior vena cava thrombosis - Inferior vena cava thrombosis - Hepatic vein thrombosis - Mesenteric vein thrombosis - Pulmonary embolism RESPIRATORY - Recurrent infections (associated with hypogammaglobulinemia) Lung - Pneumonia ABDOMEN - Abdominal pain - Ascites (in some patients) Liver - Hepatic vein thrombosis - Hepatomegaly - Budd-Chiari syndrome (in some patients) Gastrointestinal - Vomiting - Diarrhea - Malabsorption - Mucosal ulcers - Mucosal nodularity - Intestinal obstruction - Marked lymphangiectasia SKELETAL Hands - Clubbing of fingers SKIN, NAILS, & HAIR Skin - Generalized edema - Edema of face - Edema of extremities ENDOCRINE FEATURES - Hypothyroidism, subclinical HEMATOLOGY - Anemia - Thrombocytosis (in some patients) IMMUNOLOGY - Hypogammaglobulinemia LABORATORY ABNORMALITIES - Hypoproteinemia - Hypoalbuminemia - Micronutrient deficiencies MISCELLANEOUS - Variable features may be present - Recurrent thrombotic events in some patients MOLECULAR BASIS - Caused by mutation in the CD55 antigen gene (CD55, 125240.0004 ) ▲ Close
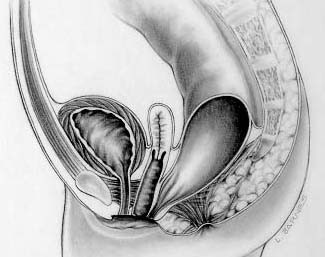
Overview A rectovaginal fistula is a connection that should not exist between the lower part of the large intestine — the rectum or anus — and the vagina. Bowel contents can leak through the fistula, allowing gas or stool to pass through the vagina. A rectovaginal fistula may result from: Injury during childbirth. Crohn's disease or other inflammatory bowel disease. Radiation treatment or cancer in the pelvic area. Complication after surgery in the pelvic area. Complication from diverticulitis, an infection of small, bulging pouches in the digestive tract.
Familial adenomatous polyposis. This rare, inherited syndrome causes certain cells on the stomach's inner lining to form a type of polyp called fundic gland polyp. When associated with this syndrome, fundic gland polyps are removed because they can become cancerous. ... Familial adenomatous polyposis. This rare, inherited syndrome increases the risk of colon cancer and other conditions, including stomach polyps.
Clinical Features Dos Santos and de Magalhaes (1980) described a family in which 10 members of 3 generations had multiple polyposis, with adenocarcinomatous propensities, limited to the stomach. No male-to-male transmission was observed. Seruca et al. (1991) restudied the family reported by dos Santos and de Magalhaes (1980) and added an observation of male-to-male transmission. Contrary to the original description, the histology of the gastric polyps present in the affected members revealed hyperplastic features and no adenomatous changes. Severe psoriasis was present in some members of the family as a probable coincidental disorder. Inheritance Based on the observation of male-to-male transmission of gastric polyposis in the family originally reported by Dos Santos and de Magalhaes (1980), Seruca et al. (1991) suggested autosomal dominant inheritance.
External links [ edit ] Classification D MeSH : D039682 External resources eMedicine : article/1082199 v t e HIV / AIDS topics HIV/AIDS HIV HIV Lentivirus structure and genome subtypes CDC classification disease progression rates HIV/AIDS diagnosis management pathophysiology prevention research vaccination PrEP WHO disease staging system for HIV infection and disease Children Teens / Adults Countries by AIDS prevalence rate Conditions Signs and symptoms AIDS-defining clinical condition Diffuse infiltrative lymphocytosis syndrome Lipodystrophy Nephropathy Neurocognitive disorders Pruritus Superinfection Tuberculosis co-infection HIV Drug Resistance Database Innate resistance to HIV Serostatus HIV-positive people Nutrition Pregnancy History History Epidemiology Multiple sex partners Timeline AIDS Museum Timothy Ray Brown Women and HIV/AIDS Social AIDS orphan Catholic Church and HIV/AIDS Circumcision and HIV Criminal transmission Discrimination against people Economic impact Cost of treatment HIV-affected community HIV/AIDS activism HIV/AIDS denialism Red ribbon Safe sex Sex education List of HIV-positive people People With AIDS Self-Empowerment Movement HIV/AIDS in the porn industry Culture Discredited HIV/AIDS origins theories International AIDS Conference International AIDS Society Joint United Nations Programme on HIV/AIDS (UNAIDS) Media portrayal of HIV/AIDS Misconceptions about HIV/AIDS President's Emergency Plan for AIDS Relief (PEPFAR) The SING Campaign Solidays Treatment Action Campaign World AIDS Day YAA/Youthforce "Free Me" Larry Kramer Gay Men's Health Crisis ACT UP Silence=Death Project HIV/AIDS pandemic by region / country Africa Angola Benin Botswana Democratic Republic of the Congo Egypt Eswatini Ethiopia Ghana Guinea Côte d'Ivoire (Ivory Coast) Kenya Lesotho Madagascar Malawi Mali Mozambique Namibia Niger Nigeria Rwanda Senegal Tanzania South Africa Uganda Zambia Zimbabwe North America Canada Mexico El Salvador Guatemala Honduras Nicaragua United States New York City Caribbean Haiti Jamaica Dominican Republic South America Bolivia Brazil Colombia Guyana Peru Asia Afghanistan Armenia Azerbaijan Bahrain Bangladesh Bhutan Cambodia China (PRC) ( Yunnan ) East Timor India Indonesia Iran Iraq Japan Jordan North Korea Laos Malaysia Myanmar (Burma) Nepal Pakistan Philippines Saudi Arabia Sri Lanka Taiwan (ROC) Thailand United Arab Emirates Turkey Vietnam Europe United Kingdom Russia Ukraine Oceania Australia New Zealand Papua New Guinea List of countries by HIV/AIDS adult prevalence rate List of HIV/AIDS cases and deaths registered by region v t e Disorders of subcutaneous fat Panniculitis Lobular without vasculitis Cold Cytophagic histiocytic Factitial Gouty Pancreatic Traumatic needle-shaped clefts Subcutaneous fat necrosis of the newborn Sclerema neonatorum Post-steroid panniculitis Lipodermatosclerosis Weber–Christian disease Lupus erythematosus panniculitis Sclerosing lipogranuloma with vasculitis: Nodular vasculitis / Erythema induratum Septal without vasculitis: Alpha-1 antitrypsin deficiency panniculitis Erythema nodosum Acute Chronic with vasculitis: Superficial thrombophlebitis Lipodystrophy Acquired generalized: Acquired generalized lipodystrophy partial: Acquired partial lipodystrophy Centrifugal abdominal lipodystrophy HIV-associated lipodystrophy Lipoatrophia annularis localized: Localized lipodystrophy Congenital Congenital generalized lipodystrophy Familial partial lipodystrophy Marfanoid–progeroid–lipodystrophy syndrome Poland syndrome
Triosephosphate isomerase (TPI) deficiency is a severe autosomal recessive inherited multisystem disorder of glycolytic metabolism characterized by hemolytic anemia and neurodegeneration. Epidemiology Prevalence of TPI deficiency is unknown and less than 50 cases have been reported in the literature. The frequency of heterozygosity was estimated at 0.4-1% among Caucasians and Asians, and 4% among African Americans. These high values suggest that homozygosity is often lethal in utero. Frequent miscarriages in the affected families support this view. Clinical description TPI deficiency is a congenital disease.
PAFAH1B1 -associated lissencephaly includes Miller-Dieker syndrome (MDS), isolated lissencephaly sequence (ILS), and (rarely) subcortical band heterotopia (SBH). ... Clinical Characteristics Clinical Description The phenotypes associated with mutation of PAFAH1B1 comprise a spectrum of severity that can be separated into Miller-Dieker syndrome (MDS), posterior predominant ILS (or isolated lissencephaly) and, on rare occasion, posterior predominant subcortical band heterotopia (SBH). ... Two children with Miller-Dieker syndrome showing typical facial features Photographs obtained with consent of the families. ... Children with lissencephaly have epileptic encephalopathies that typically evolve from infantile spasms (West syndrome) to Lennox-Gastaut syndrome of mixed epilepsy with a slow spike and wave pattern on EEG. ... The cobblestone cortical malformation (lissencephaly) syndromes (Walker-Warburg syndrome, muscle-eye-brain disease, and Fukuyama congenital muscular dystrophy) differ clinically in a number of ways, including the frequent presence of hydrocephalus and cerebellar hypoplasia, multiple different eye anomalies, and congenital muscular dystrophy manifest by hypotonia and elevated serum creatine kinase concentrations.
Since the phenotype of MAP (as described in Suggestive Findings) overlaps with a number of other hereditary polyposis and colorectal cancer syndromes (see Differential Diagnosis), many individuals with MAP will likely be diagnosed using a multigene panel. ... Of note, eight of 17 persons with MAP had one or more hyperplastic colonic polyps and/or sessile serrated adenomas; findings in three of these eight individuals met criteria for the serrated polyposis syndrome (see Differential Diagnosis) [Boparai et al 2008]. ... In a minority (0%-3.7%) of affected individuals, biallelic germline MUTYH pathogenic variants have been found in the index case of a family with a phenotype suggestive of Lynch syndrome [Nielsen et al 2011, Castillejo et al 2014]. ... Disorders to Consider in the Differential Diagnosis of MUTYH Polyposis View in own window Cancer Susceptibility Syndrome Gene(s) / Genetic Mechanism MOI Polyps / Colon Cancer Associated Malignancies Other Features / Comments APC -associated polyposis conditions APC AD Attenuated FAP: 0-100 colonic polyps CRC risk: 70% FAP: 100 colonic polyps Upper GI polyps CRC risk: 100% Small bowel Pancreatic Thyroid Liver Brain Bile duct Gastric CHRPE Osteomas Supernumerary or missing teeth Cutaneous lesions Desmoid tumors NTHL1- associated polyposis NTHL1 AR 8-50 adenomatous colonic polyps Duodenal adenomas CRC in 16/29 individuals Extracolonic cancer in 12/29 individuals: Uterine Duodenal Breast Premalignant endometrial lesions 2nd most common AR form of colon cancer & polyps after MAP 1 Lynch syndrome (hereditary non-polyposis colon cancer) MLH1 MSH2 MSH6 PMS2 EPCAM AD 4% develop ≥10 polyps Colon tumors often MSI+ CRC risk: 52%-82% Uterine Ovarian Small bowel Gastric Urinary tract Skin Brain Hepatobiliary tract Pancreas Prostate Peutz-Jeghers syndrome STK11 AD GI hamartomatous polyps Polyps most often in small bowel Adenomatous colonic polyps can occur. CRC risk: 39% Gastric Breast Ovarian Small bowel Pancreas Cervix Uterine Lung Testicular Ovarian sex cord tumors w/annular tubules Dk-brown to dk-blue melanocytic macules (fade w/age) Juvenile polyposis syndrome BMPR1A SMAD4 AD Hamartomatous (juvenile) polyps in small bowel, stomach, colon, & rectum CRC risk: 38.7% Gastric Upper GI tract Pancreas Hereditary hemorrhagic telangiectasia ( SMAD4- related) PTEN hamartoma tumor syndrome PTEN AD Multiple hamartomatous & mixed polyps in GI tract CRC risk: 9% Breast Thyroid Uterine Renal Brain Melanoma Thyroid disease/nodules Uterine fibroids Macrocephaly Lipomas Mucocutaneous lesions Pigmented macules of glans penis Hamartomatous overgrowth of tissues Connective tissue nevi Epidermal nevi Hyperostoses Hereditary mixed polyposis syndrome (OMIM 610069, 601228) BMPR1A GREM1 dup 15q13-q14 2 AD Adenomatous polyps, juvenile polyps, hyperplastic polyps, & polyps containing mixed histology ↑ CRC risk 3 Desmoid tumor, prostate cancer, & duodenal adenocarcinoma reported in 1 individual 4 Rare condition (few families) Sessile serrated polyposis syndrome (OMIM 617108) RNF43 AD Sessile serrated polyps, serrated adenomas, or hyperplastic polyps of GI tract CRC risk: ≤54% Unknown (various cancers reported in untested family members) Rare condition (few individuals) AD = autosomal dominant; AR = autosomal recessive; CHRPE = congenital hypertrophy of the retinal pigment epithelium; CRC = colorectal cancer; FAP = familial adenomatous polyposis; MAP = MUTYH polyposis; MOI = mode of inheritance; MSI = microsatellite instability 1.
The cerebral creatine deficiency syndromes (CCDS), inborn errors of creatine metabolism, include the two creatine biosynthesis disorders, guanidinoacetate methyltransferase (GAMT) deficiency and L-arginine:glycine amidinotransferase (AGAT) deficiency, and the creatine transporter (CRTR) deficiency. ... Algorithm for diagnosis of the cerebral creatine deficiency syndromes. Note: Urinary creatine/creatinine ratio and creatine uptake studies in cultured skin fibroblasts are often not informative in females with SLC6A8 deficiency; hence, molecular genetic (more...) ... Two affected males with mild cardiomyopathy were reported [Puusepp et al 2010]. One affected male had long QT syndrome [van de Kamp et al 2013a]. Medical concerns in adulthood. ... Boenzi et al [2012] measured plasma creatine levels in individuals with ornithine transcarbamylase (OTC), argininosuccinate synthetase (ASS), and argininosuccinate lyase (ASL) deficiencies; hyperammonemia, hyperornithinemia, homocitrullinuria (HHH) syndrome; and lysinuric protein intolerance (LPI). Individuals with OTC and ASS deficiencies and HHH syndrome showed significant reduction of plasma creatine concentration, whereas individuals with ASL deficiency and LPI had high plasma creatine levels.
., dehydration), low blood pressure , heart failure (leading to cardiorenal syndrome ), hepatorenal syndrome in the context of liver cirrhosis, and local changes to the blood vessels supplying the kidney. ... Other causes of intrinsic AKI are rhabdomyolysis and tumor lysis syndrome . [10] Certain medication classes such as calcineurin inhibitors (e.g., tacrolimus ) can also directly damage the tubular cells of the kidney and result in a form of intrinsic AKI. ... Indications for kidney biopsy in the setting of AKI include the following: [15] Unexplained AKI, in a patient with two non-obstructed normal sized kidneys AKI in the presence of the nephritic syndrome Systemic disease associated with AKI Kidney transplant dysfunction In medical imaging , the acute changes in the kidney are often examined with renal ultrasonography as the first-line modality, where CT scan and magnetic resonance imaging (MRI) are used for the follow-up examinations and when US fails to demonstrate abnormalities. ... External links [ edit ] Classification D ICD - 10 : N17 ICD - 9-CM : 584 MeSH : D058186 DiseasesDB : 11263 External resources MedlinePlus : 000501 eMedicine : med/1595 Patient UK : Acute kidney injury v t e Kidney disease Glomerular disease See Template:Glomerular disease Tubules Renal tubular acidosis proximal distal Acute tubular necrosis Genetic Fanconi syndrome Bartter syndrome Gitelman syndrome Liddle's syndrome Interstitium Interstitial nephritis Pyelonephritis Balkan endemic nephropathy Vascular Renal artery stenosis Renal ischemia Hypertensive nephropathy Renovascular hypertension Renal cortical necrosis General syndromes Nephritis Nephrosis Renal failure Acute renal failure Chronic kidney disease Uremia Other Analgesic nephropathy Renal osteodystrophy Nephroptosis Abderhalden–Kaufmann–Lignac syndrome Diabetes insipidus Nephrogenic Renal papilla Renal papillary necrosis Major calyx / pelvis Hydronephrosis Pyonephrosis Reflux nephropathy v t e Organ failure General Heart failure Respiratory failure Liver failure Acute Chronic Renal failure Acute Chronic Encephalopathy Multiple Multiple organ dysfunction syndrome
Review of the psychiatric and criminal phenomena "Malingering by Proxy" or "Commando Syndrome" occurring amongst Apathetic Refugee Children in Sweden . ^ Jackson, Thomas. ... "Asylum seeking children with resignation syndrome - Trauma, culture and asylum process" . ... Retrieved 15 July 2019 . ^ Jackson, Thomas. "Jackson-Mal-BP-Syndrom | Jackson Jagar Journalister (Jackson hunts reporters)" . ... Retrieved 15 July 2019 . ^ Nunn, Kenneth P.; Lask, Bryan; Owen, Isabel (2013). "Pervasive refusal syndrome (PRS) 21 years on: a re-conceptualisation and a renaming" . ... Retrieved 15 July 2019 . ^ Radio, Sveriges (Bill Schiler). "Apathetic Children's Syndrome On The Rise - Radio Sweden" . sverigesradio.se (The apathetic refugee children).
Retrieved 2018-10-21 . ^ a b Wessely, Simon (1987). "Mass hysteria: two syndromes?" . Psychological Medicine . 17 (1): 109–20. doi : 10.1017/s0033291700013027 . ... Web. 17 Dec. 2009. ^ Ali-Gombe, A. et al. "Mass hysteria: one syndrome or two?" British Journal of Psychiatry 1997; 170 387–88. ... "U.S. diplomats in Cuba have unusual brain syndrome, but there's no proof they were attacked, study says" . ... General references [ edit ] Ali-Gombe, A. et al. "Mass hysteria: one syndrome or two?" British Journal of Psychiatry 1997; 170 387–78. ... Wessely, Simon (1987). "Mass hysteria: two syndromes?" . Psychological Medicine . 17 (1): 109–20. doi : 10.1017/s0033291700013027 .