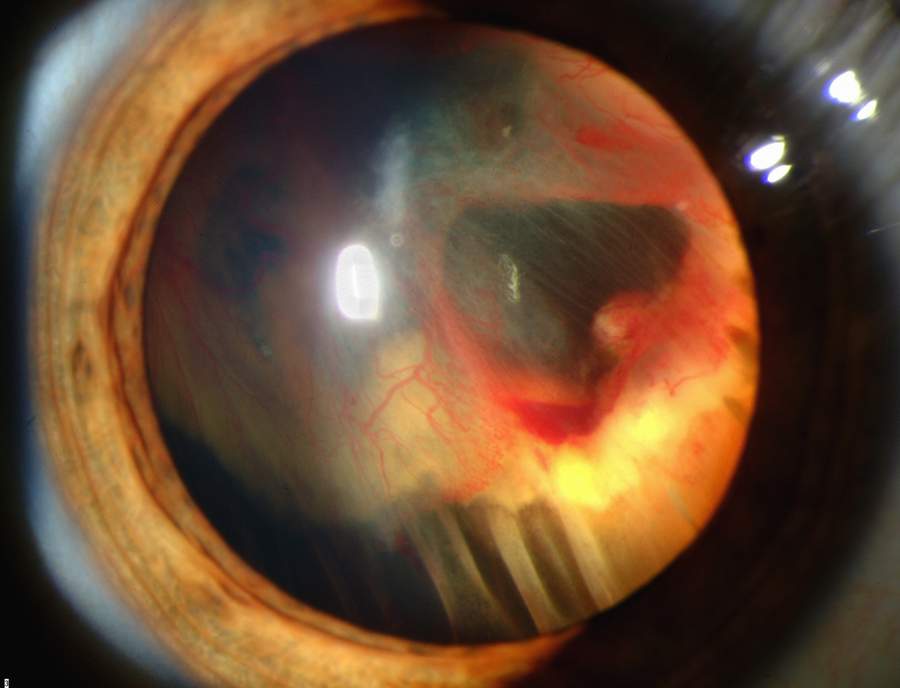
Overview Retinal detachment describes an emergency situation in which a thin layer of tissue (the retina) at the back of the eye pulls away from its normal position. Retinal detachment separates the retinal cells from the layer of blood vessels that provides oxygen and nourishment to the eye. The longer retinal detachment goes untreated, the greater your risk of permanent vision loss in the affected eye. Warning signs of retinal detachment may include one or all of the following: reduced vision and the sudden appearance of floaters and flashes of light. Contacting an eye specialist (ophthalmologist) right away can help save your vision.
Clinical Features Boyer et al. (1975) described a woman with the Raynaud phenomenon, sclerodactyly, and telangiectasia (incomplete CRST syndrome; see 181750). X-rays showed no subcutaneous calcification, but the interosseous membrane between the radius and ulna was calcified. ... They considered the association of C7 deficiency and CRST syndrome to be coincidental, mainly because 2 other cases of CRST had high C7 levels.
Clinical Features Doman et al. (1990) used the designation spondyloepiphyseal dysplasia (SED) of Maroteaux for a form of spondyloepiphyseal dysplasia with manifestations limited to the musculoskeletal system and with other features distinguishing it from Morquio syndrome of any type (253000, 253010, 252300), X-linked SED tarda (313400), brachyolmia (113500, 271530, 271630), and spondylometaphyseal dysplasia of Kozlowski (184252). ... The pelvic inlet is described as having a champagne-glass configuration, unlike the wine-glass-like configuration of the pelvic inlet in patients who have Morquio syndrome. Odontoid hypoplasia was not found.
Spondyloepiphyseal dysplasia, Maroteaux type is a very rare type of spondyloepiphyseal dysplasia (see this term) described in fewer than 10 patients to date and characterized clinically by dysplastic epiphyses, short stature appearing in infancy, short neck, short and stubby hands and feet, scoliosis, genu valgum, abnormal pelvis, osteoporosis and osteoarthritis.
Spondyloepiphyseal dysplasia (SED) Maroteaux type is a rare skeletal dysplasia that is characterized by short stature beginning in infancy, short, stubby hands and feet, and genu valgum (knock knees). In addition to these physical characteristics, individuals with SED Maroteaux type have some common radiographic findings, including platyspondyly (flattened vertebral bodies in the spine), abnormalities of the pelvis and severe brachydactyly (short fingers and toes). Intelligence is generally normal and there is no clouding of the cornea, which distinguishes SED Maroteaux type from other forms of spondyloepiphyseal dysplasia. SED Maroteaux type is caused by mutations in the TRPV4 gene and is inherited any an autosomal dominant fashion.
Description The term 'brachyolmia' was coined to designate a bone dysplasia characterized clinically by short trunk dwarfism and radiographically by generalized platyspondyly without significant long bone abnormalities. The Maroteaux type of brachyolmia is an autosomal recessive form in which there is rounding of the anterior and posterior vertebral borders, with less elongation on lateral view and less lateral extension on anteroposterior view than is seen in the Hobaek type of brachyolmia (271530). Maroteaux brachyolmia may also be associated with precocious calcification of the falx cerebri, and minor facial anomalies (summary by Shohat et al., 1989). For a discussion of genetic heterogeneity of brachyolmia, see 271530. Clinical Features Shohat et al. (1989) reported 4 families with the Maroteaux type of brachyolmia, including 2 brothers whose Israeli parents were first cousins, suggesting autosomal recessive inheritance.
A relatively mild form of brachyolmia, a group of rare genetic skeletal disorders, characterized by short trunk/short stature, generalized platyspondyly and rounding of vertebral bodies. It remains unknown whether the phenotype represents a single disease entity or a heterogeneous group of mild skeletal dysplasias. Epidemiology The prevalence of this form of brachyolmia is not known. About 10 cases have been reported. Clinical description Patients with brachyolmia, Maroteaux type are reported to have short stature/short trunk, scoliosis and generalized platyspondyly with rounding of the anterior and posterior vertebral bodies. The vertebral bodies show less elongation compared to those in patients with other types of the disorder (autosomal recessive brachyolmia, Hobaek/Toledo type, autosomal dominant brachyolmia; see these terms).
Puffenberger et al. (2012) noted some phenotypic similarities to Angelman syndrome (AS; 105830). Harlalka et al. (2013) reported 15 additional Amish individuals, aged 11 months to 39 years, with this severe neurodevelopmental disorder. ... Puffenberger et al. (2012) noted that this pathophysiologic mechanism is similar to that thought to underlie Angelman syndrome (AS; 105830), one of a group of neurodevelopmental disorders mapping to chromosome 15q11-q13, which results from loss of UBE3A (601623).
Developmental delay with autism spectrum disorder and gait instability is a rare, genetic, neurological disorder characterized by infant hypotonia and feeding difficulties, global development delay, mild to moderated intellectual disability, delayed independent ambulation, broad-based gait with arms upheld and flexed at the elbow with brisk walking or running, and limited language skills. Behavior patterns are highly variable and range from sociable and affectionate to autistic behavior.
It turned out that these patients with hepatic steatosis had nonketotic C6-C10-dicarboxylic aciduria with episodes of a Reye-like syndrome and profound hypoglycemia. The conclusion was based on findings in a third affected sib still living, aged 7 at the time of report. After a respiratory infection at 13 months she became semicomatose, developing hepatomegaly and mild hypoglycemia. A similar but less severe Reye-syndrome-like episode occurred at 6 years.
He called this disorder the black lock-albinism-deafness syndrome (BADS). O'Doherty and Gorlin (1988) presented photographs of a striking patient who had a nearly normal iris pigmentation but white hair and white eyebrows and eyelashes with scattered black tufts resembling ermine (the ermine is born with brown fur which changes at about age 9 months, when it grows its characteristic winter coat for camouflage in a snowy environment, but with the black tip of its tail always preserved). ... O'Doherty and Gorlin (1988) referred to descriptions of oculocutaneous albinism with black locks and congenital sensorineural hearing loss (BADS syndrome) occurring in the brother and sister described by Witkop (1979).
A rare deafness characterized by the association of bilateral sensorineural hearing loss and white hair with scattered black tufts, as well as skin areas of hyper- and hypopigmentation. Additional reported features include global developmental delay and moderate intellectual disability, growth retardation, microcephaly, hypotonia, mild dysmorphic facial features (deeply set eyes, broad nasal bridge, slight bowing of the upper lip), retinal depigmentation, anomalies of the fingers and toes, and white matter abnormalities on brain imaging.
Brain MRI showed bilateral lesions in the thalami, brainstem, and medulla spinalis, consistent with a diagnosis of Leigh syndrome (256000). By the time of her death at age 8 years, she was hypermobile with ataxia and used a wheelchair. ... Antonicka et al. (2010) noted that the disease progression in these patients was slower than that seen in patients with classic Leigh syndrome. Biochemical Features By laboratory studies of fibroblasts isolated from patients with a complex neurologic disorder, Antonicka et al. (2010) found severe assembly defects of mitochondrial respiratory complexes I, IV, and V, with a milder defect in the assembly of complex III.
Combined oxidative phosphorylation defect type 7 is a rare mitochondrial disease due to a defect in mitochondrial protein synthesis characterized by a variable phenotype that includes onset in infancy or early childhood of failure to thrive and psychomotor regression (after initial normal development), as well as ocular manifestations (such as ptosis, nystagmus, optic atrophy, ophthalmoplegia and reduced vision). Additional manifestations include bulbar paresis with facial weakness, hypotonia, difficulty chewing, dysphagia, mild dysarthria, ataxia, global muscle atrophy, and areflexia. It has a relatively slow disease progression with patients often living into the third decade of life.
The 2 Mexican patients had mild mental retardation, delayed walking at about 3 years, cataracts, strabismus, ptosis, and cardiac anomalies. One had long QT syndrome (see LQT1, 192500) and the other had left ventricular dilatation. ... INHERITANCE - Autosomal recessive HEAD & NECK Head - Microcephaly - Poor head control Face - Myopathic face Eyes - Cataracts (in some patients) - Strabismus (in some patients) - Ptosis (in some patients) - Nystagmus (in some patients) CARDIOVASCULAR Heart - Long QT syndrome (1 patient) - Left ventricular dilatation (1 patient) ABDOMEN Gastrointestinal - Feeding difficulties SKELETAL - Contractures (in some patients) MUSCLE, SOFT TISSUES - Muscular dystrophy - Hypotonia - Muscle weakness, severe - Generalized muscle wasting - Increased muscle tone (early in life) - Dystrophic features seen on muscle biopsy - Hypoglycosylation of alpha-dystroglycan seen on muscle biopsy NEUROLOGIC Central Nervous System - Delayed psychomotor development - Mental retardation, mild to severe - Absent speech - Delayed or absent independent walking - Seizures (in some patients) - Cerebellar hypoplasia (in some patients) PRENATAL MANIFESTATIONS Movement - Decreased fetal movements LABORATORY ABNORMALITIES - Increased serum creatine kinase MISCELLANEOUS - Onset at birth or in early infancy - Variable severity MOLECULAR BASIS - Caused by mutation in the GDP-mannose pyrophosphorylase B gene (GMPPB, 615320.0004 ) ▲ Close
The combination of an accessory pathway that causes pre-excitation with arrhythmias is known as Wolff-Parkinson-White syndrome . [2] Accessory pathways are often diagnosed using an electrocardiogram, but characterisation and location of the pathway may require an electrophysiological study . ... OCLC 938434294 . ^ Bhatia, Atul; Sra, Jasbir; Akhtar, Masood (March 2016). "Preexcitation Syndromes". Current Problems in Cardiology . 41 (3): 99–137. doi : 10.1016/j.cpcardiol.2015.11.002 .
Cholinergic crisis Other names Cholinergic toxicity, cholinergic poisoning, SLUDGE syndrome A cholinergic crisis is an over-stimulation at a neuromuscular junction due to an excess of acetylcholine (ACh), [1] as a result of the inactivity of the AChE enzyme , which normally breaks down acetylcholine. ... It is useful to remember some of the symptoms of increased cholinergic stimulation that include: Salivation : stimulation of the salivary glands Lacrimation : stimulation of the lacrimal glands (tearing) Urination : relaxation of the internal sphincter muscle of urethra , and contraction of the detrusor muscles Defecation Gastrointestinal distress : Smooth muscle tone changes causing gastrointestinal problems , including cramping Emesis : Vomiting [2] Miosis [3] constriction of the pupils of the eye via stimulation of the pupillary constrictor muscles Muscle spasm : stimulation of skeletal muscle (due to nicotinic acetylcholine receptor stimulation) Cause [ edit ] Cholinergic crisis, sometimes known by the mnemonic "SLUDGE syndrome" ( Salivation, Lacrimation, Urination, Defecation, Gastrointestinal Distress and Emesis), [4] can be a consequence of: Contamination with - or excessive exposure to - certain chemicals including: nerve agents , ( e.g. sarin , VX , Novichok agents ). organophosphorus insecticides ( e.g. parathion ) nicotine poisoning can also present with similar symptoms, as it also involves excessive parasympathetic stimulation. [5] Ingestion of certain poisonous fungi (particularly the muscarine -containing members of the genera Amanita , Inocybe and Clitocybe ).
Adrenocortical carcinoma , cancer of the adrenal cortex Adrenal incidentaloma , an adrenal tumor (of any type) discovered accidentally during a scan which performed for an unrelated reason Pheochromocytoma , a catecholamine -producing tumor of the adrenal medulla, which may or may not be cancerous Hereditary disorders associated with adrenal tumors [ edit ] Von Hippel–Lindau disease , a mutation of the VHL1 tumor-suppression gene associated with many types of tumor, including pheochromocytoma Multiple Endocrine Neoplasia , a family of syndromes in which genetic abnormalities contribute to the development of endocrine tumors Notable people with adrenal gland disorders [ edit ] John F. ... Cushing's disease , a disorder in which cortisol levels are abnormally high Hyperaldosteronism (including Conn's syndrome ), a condition in which aldosterone is over-produced Hypoaldosteronism , a condition in which aldosterone is under-produced References [ edit ] ^ "Overview of the Adrenal Glands: Adrenal Gland Disorders: Merck Manual Home Health Handbook" .
Molecular pathology [ edit ] Michel aplasia is associated with LAMM syndrome(labyrinthine aplasia, microtia and microdontia), which is caused by mutation FGF3 gene on chromosome 11q13 which encodes fibroblast growth factor 3. ... As michel's aplasia is associated with LAMM syndrome there will be Microtia and microdontia present(small sized teeth).
Hypersalivation also often precedes emesis (vomiting), where it accompanies nausea (a feeling of needing to vomit). [5] Contents 1 Causes 1.1 Excessive production 1.2 Decreased clearance 2 Treatment 3 References 4 External links Causes [ edit ] Excessive production [ edit ] Conditions that can cause saliva overproduction include: [4] Rabies Pellagra (niacin or Vitamin B3 deficiency) [6] Gastroesophageal reflux disease , in such cases specifically called a water brash (a loosely defined layman term), and is characterized by a sour fluid or almost tasteless saliva in the mouth [7] Gastroparesis (main symptoms are nausea, vomiting, and reflux) Pregnancy Excessive starch intake Anxiety (common sign of separation anxiety in dogs ) Pancreatitis Liver disease Serotonin syndrome Mouth ulcers [ medical citation needed ] Oral infections Sjögren syndrome (an early symptom in some patients) [8] Medications that can cause overproduction of saliva include: [4] aripiprazole clozapine pilocarpine ketamine potassium chlorate risperidone pyridostigmine Substances that can cause hypersalivation include: [4] mercury copper organophosphates ( insecticide ) arsenic nicotine thallium Decreased clearance [ edit ] Causes of decreased clearance of saliva include: [4] Infections such as tonsillitis , retropharyngeal and peritonsillar abscesses , epiglottitis and mumps .
Chronic actinic dermatitis (CAD) is an immunologically mediated photodermatosis usually observed in temperate climates and that typically develops in middle-aged to elderly males. CAD is characterized by eczematous and often lichenified pruritic patches and confluent plaques located predominantly on sun-exposed areas with notable sparing of eyelids, skin folds, and postauricular skin. It is often accompanied by multiple contact allergies and usually occurs in a background of either atopic, contact allergic, or seborrheic dermatitis, although it can occur de novo . Resolution of photosensitivity is reported in up to 50% of individuals after 15 years or more, with contact allergies persisting.
Prognosis [ edit ] No adverse effects or consequences of melanosis coli have been identified. [4] Relation to true melanoses [ edit ] The condition is unrelated to true melanoses, such as Peutz–Jeghers syndrome and smoker's melanosis . [5] Peutz–Jeghers syndrome causes pigmentation of the skin and mucous surfaces with melanin , and polyps in the digestive tract .