Amniotic membrane transplantation can also be fixated to the sclera using sutures, or glue adhesive. [26] [27] [28] Amniotic membrane by itself does not provide an acceptable recurrence rate. [29] References [ edit ] ^ Tollefsbol, Trygve (2016). ... Ophthalmic Epidemiology . 6 (3): 219–28. doi : 10.1076/opep.6.3.219.1504 . ... Retrieved 30 November 2012 . ^ "Pterygium: MedlinePlus Medical Encyclopedia" . medlineplus.gov . Archived from the original on 28 August 2016 . Retrieved 15 August 2016 . ^ "Pterygium Workup: Imaging Studies, Procedures" . emedicine.medscape.com .
They have been found efficient in alleviating hot flashes. [23] On 28 June 2013 FDA approved Brisdelle (low-dose paroxetine mesylate) for the treatment of moderate-to-severe vasomotor symptoms (e.g. hot flashes and night sweats) associated with menopause. ... In a large double-blinded randomized controlled trial, reduction in hot flashes was not statistically significant but showed a strong trend towards improvement. [28] Lack of statistical significance suggests future research, but does not meet the scientific bar for ginseng to be deemed effective. ... S2CID 22844064 . ^ Food and Drug Administration (28 June 2013). "FDA NEWS RELEASE: FDA approves the first non-hormonal treatment for hot flashes associated with menopause" .
Overview A hot flash is the sudden feeling of warmth in the upper body, which is usually most intense over the face, neck and chest. Your skin might redden, as if you're blushing. A hot flash can also cause sweating. If you lose too much body heat, you might feel chilled afterward. Night sweats are hot flashes that happen at night, and they may disrupt your sleep. Although other medical conditions can cause them, hot flashes most commonly are due to menopause — the time when menstrual periods become irregular and eventually stop. In fact, hot flashes are the most common symptom of the menopausal transition.
NHLBI . 22 June 2016. Archived from the original on 28 July 2016 . Retrieved 31 August 2016 . ^ a b c d e "What Causes Cardiomyopathy?" ... - NHLBI, NIH" . www.nhlbi.nih.gov . Archived from the original on 28 July 2016 . Retrieved 25 July 2016 . ^ Lakdawala, NK; Stevenson, LW; Loscalzo, J (2015). ... "Viral myocarditis" . Current Opinion in Rheumatology . 28 (4): 383–9. doi : 10.1097/BOR.0000000000000303 .
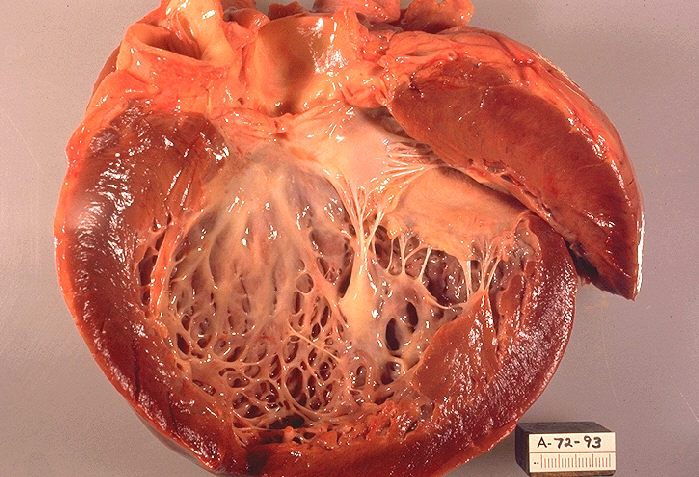
Overview Cardiomyopathy (kahr-dee-o-my-OP-uh-thee) is a disease of the heart muscle that makes it harder for the heart to pump blood to the rest of the body. Cardiomyopathy can lead to heart failure. The main types of cardiomyopathy include dilated, hypertrophic and restrictive cardiomyopathy. Treatment — which might include medications, surgically implanted devices, heart surgery or, in severe cases, a heart transplant — depends on the type of cardiomyopathy and how serious it is. Types Symptoms There might be no signs or symptoms in the early stages of cardiomyopathy. But as the condition advances, signs and symptoms usually appear, including: Breathlessness with activity or even at rest Swelling of the legs, ankles and feet Bloating of the abdomen due to fluid buildup Cough while lying down Difficulty lying flat to sleep Fatigue Heartbeats that feel rapid, pounding or fluttering Chest discomfort or pressure Dizziness, lightheadedness and fainting Signs and symptoms tend to get worse unless treated.
These data suggests drugs targeting mutant BRAF as potential novel therapies for ameloblastoma. [25] SMO mutations lead to the activation of the hedgehog pathway giving similar results as V600E but is less frequently seen. [8] 55% of SMO mutations are found in the maxilla. [7] Evidence shows that suppression of matrix metalloproteinase-2 may inhibit the local invasiveness of ameloblastoma, however, this was only demonstrated in vitro . [26] There is also some research suggesting that α 5 β 1 integrin may participate in the local invasiveness of ameloblastomas. [27] Epidemiology [ edit ] People with African heritage have been shown to have a higher incidence compared to Caucasians, with the site often being in the midline of the mandible. [10] The annual incidence rates per million for ameloblastomas are 1.96, 1.20, 0.18 and 0.44 for black males, black females, white males and white females respectively. [28] Ameloblastomas account for about one percent of all oral tumors [16] and about 18% of odontogenic tumors. [29] Men and women are equally affected, though women average four years younger than men when tumors first occur, and tumors run larger in females. [1] See also [ edit ] Ameloblastic fibroma Bone grafting Epithelial cell rests of Malassez List of cutaneous conditions Matrix Metalloproteinase-2 Tooth development and Odontogenesis References [ edit ] ^ a b c d e f g h Reichart PA, Philipsen HP, Sonner S (March 1995). ... PMID 28984343 . ^ a b c d e f g h i j k l m n o p q W., Odell, E. (2017-06-28). Cawson's essentials of oral pathology and oral medicine . ... "Recurrent ameloblastoma in an autogenous bone graft after 28 years: a case report". The New Zealand Dental Journal . 91 (403): 12–3.
A rare, benign, slow-growing odontologic tumor located in the mandible, and on occasion the maxilla, characterized by painless, variable-sized jaw swelling, which if left untreated may lead to a grotesque facial appearance. Occasionally, paresthesias, tooth displacement and adjacent root resorption may be associated. Local invasion is frequently observed, but malignant transformation and metastasis are not common.
Overview Ameloblastoma is a rare, noncancerous (benign) tumor that develops most often in the jaw near the molars. Ameloblastoma begins in the cells that form the protective enamel lining on your teeth. The most common type of ameloblastoma is aggressive, forming a large tumor and growing into the jawbone. Treatment may include surgery and radiation. In some cases, reconstruction may be necessary to restore your teeth, jaw and facial appearance. Some types of ameloblastoma are less aggressive. Though ameloblastoma is most often diagnosed in adults in their 30s through 60s, ameloblastoma can occur in children and young adults.
Ameloblastoma is a rare, noncancerous (benign) tumor that typically develops in the jaw near the molars. It originates in the cells that form the enamel that protects your teeth. The condition most often occurs in adults in their 30s and 40s, though it can occur at any age. In many cases, the first sign is painless swelling in the jaw. While it can be very aggressive, these tumors are rarely found outside of the jaw. Treatment is complete surgical removal of the affected tissue.
Molecular Genetics Von Willebrand Disease Type 2A Mutations causing the enhanced proteolysis phenotype lie within or near domain A2 (exon 28) of the VWF gene, which is the site of the ADAMTS13 (604134) cleavage sequence between residues tyr1605 and met1606. ... In patients with VWD type 2B, Randi et al. (1991) identified 3 different heterozygous mutations in exon 28 of the VWF gene (613160.0005-613160.0007) within the domain that interacts with platelet glycoprotein GP1BA, resulting in a loss of function. ... The region of VWF that binds to GP1BA has been localized to a peptide including amino acids 480 to 718 of the mature subunit that is encoded by exon 28. In affected members of a Swedish family (Holmberg et al., 1986) and a German family with a variant of VWD type 2B, Holmberg et al. (1993) identified a heterozygous mutation in the VWF gene (P1266L; 613160.0033).
A form of von Willebrand disease (VWD) characterized by a bleeding disorder associated with a qualitative deficiency and functional anomalies of the Willebrand factor (VWF). Depending on the type of functional abnormalities, this form is classified as type 2A, 2B, 2M or 2N. Epidemiology The subtypes of type 2 VWD account for between 20-45% of cases of VWD. Clinical description Age of onset of the bleeding anomalies varies, with earlier onset being associated with more severe VWF deficiency. Four type 2 VWD subtypes have been described: types 2A, 2B and 2M are characterized by mucocutaneous manifestations (menorrhagia, epistaxis, gastrointestinal hemorrhage etc.); type 2N is mainly characterized by post traumatic soft tissue bleedings.
The prevalence has been estimated somewhere between 1 in 75,000 and 1 in 200,000 [26] however it has been noted that the prevalence of EPP may be increasing due to a better understanding of the disease and improved diagnosis. [27] An estimated 5,000-10,000 individuals worldwide have EPP. [ medical citation needed ] EPP is considered the most common form of porphyria in children. [28] The prevalence in Sweden has been published as 1:180,000. [29] History [ edit ] Erythropoietic protoporphyria was first described in 1953 by Kosenow and Treibs [30] and completed in 1960 by Magnus et al. at the St John's Institute of Dermatology in London. [31] See also [ edit ] Xeroderma pigmentosum Porphyria cutanea tarda List of cutaneous conditions References [ edit ] ^ RESERVED, INSERM US14-- ALL RIGHTS. ... Anthony; Moreno-Otero, Ricardo (2010-09-28). "Liver disease and erythropoietic protoporphyria: a concise review" . ... "[Liver disease in erythropoietic protoporphyria]". Gastroenterología y Hepatología . 28 (10): 632–636. doi : 10.1016/s0210-5705(05)71529-6 .
Erythropoietic protoporphyria (EPP) is a type of porphyria . Porphyrias are caused by an abnormality in the heme production process. Heme is essential in enabling our blood cells to carry oxygen and in breaking down chemical compounds in the liver. Erythropoietic protoporphyria is caused by pathogenic variants (mutations) in the FECH gene which lead to an impaired activity of ferrocheletase (FECH), an important enzyme in heme production. This results in the build-up of protoporphyrin in the bone marrow, red blood cells, blood plasma, skin, and eventually liver. Build up of protoporphyrin can cause extreme sensitivity to sunlight, liver damage, abdominal pain, gallstones, and enlargement of the spleen.
A number sign (#) is used with this entry because erythropoietic protoporphyria-1 (EPP1) is caused by compound heterozygous or homozygous mutation in the gene encoding ferrochelatase (FECH; 612386) on chromosome 18q21. The disorder most often results from inheritance of a null FECH allele in trans with a low-expression FECH mutation (612386.0015) prevalent in some populations, resembling autosomal dominant inheritance with incomplete penetrance. Description Erythropoietic protoporphyria-1 is an inborn error of porphyrin metabolism caused by decreased activity of the enzyme ferrochelatase, the terminal enzyme of the heme biosynthetic pathway, which catalyzes the insertion of iron into protoporphyrin to form heme. EPP is characterized clinically by photosensitivity to visible light commencing in childhood, and biochemically by elevated red cell protoporphyrin levels (Todd, 1994). Genetic Heterogeneity of Erythropoietic Protoporphyria Also see X-linked erythropoietic protoporphyria (XLEPP; 300752), caused by mutation in the ALAS2 gene (301300) on chromosome Xp11, and EPP2 (618015), caused by mutation in the CLPX gene (615611) on chromosome 15q22.
Erythropoietic protoporphyria (EPP) is an inherited disorder of the heme metabolic pathway characterized by accumulation of protoporphyrin in blood, erythrocytes and tissues, and cutaneous manifestations of photosensitivity. Epidemiology EPP has been reported worldwide, with prevalence ranging between 1/75,000 and 1/200,000. Clinical description EPP usually manifests in early infancy upon the first exposure to sun. EPP is characterized by cutaneous manifestations of acute painful photosensitivity with erythema and edema, sometimes with petechiae, together with stinging and burning sensations without blistering, upon exposure to sunlight or artificial light (400-700 nm). These episodes have a variable severity depending on the exposure duration and may result in chronic permanent lesions on exposed skin.
. ^ a b c d Rittler, M; Paz, JE; Castilla, EE (Jun 28, 1996). "VACTERL association, epidemiologic definition and delineation". ... Further reading [ edit ] McMullen, KP; Karnes, PS; Moir, CR; Michels, VV (Jun 28, 1996). "Familial recurrence of tracheoesophageal fistula and associated malformations".
VACTERL/VATER is an association of congenital malformations typically characterized by the presence of at least three of the following: vertebral defects, anal atresia, cardiac defects, tracheo-esophageal fistula, renal anomalies, and limb abnormalities. Epidemiology Exact prevalence and incidence data are not available due to variable diagnostic criteria, but the association has been reported to occur in <1-9/100,000 infants, and the annual incidence has been reported to be 1/10,000 to 1/40,000 live births. No specific geographic distribution or predominance in certain ethnic groups has been found. Clinical description A cluster of congenital malformations is found at birth or in the first days of life and includes at least three component features: vertebral defects (60-80% of patients), commonly accompanied by rib anomalies; imperforate anus/anal atresia (55-90%); cardiac defects (40-80%); tracheo-esophageal fistula (50-80%), with or without esophageal atresia; renal anomalies (50-80%) including renal agenesis, horsehoe kidney, and cystic and/or dysplastic kidneys; and limb abnormalities (40-50%). Limb defects are classically defined as radial anomalies, including thumb aplasia/hypoplasia, and have variable degrees of severity; other types of limb anomalies have also been reported.
Chafa et al. (1989) reported 2 sibs, a 26-year-old woman and her 28-year-old brother, who had recurrent venous thrombosis since the age of 20. ... Among a cohort 109 first-degree relatives of 28 patients with genetically confirmed protein S deficiency, a low free protein S level was the most reliable predictor of a PROS1 gene defect (sensitivity 97.7%, specificity 100%).
An inherited coagulation disorder characterized by recurrent venous thrombosis symptoms due to reduced synthesis and/or activity levels of protein S. Epidemiology Prevalence of partial protein S deficiency (heterozygous individuals) is estimated at 0.16-0.21% in the general population. Prevalence of severe protein S deficiency (homozygous or compound heterozygous individuals) is unknown but is probably comparable to that of severe protein C deficiency which is estimated at 1/500,000. Men and women are equally affected. Clinical description In severe protein S deficiency, the disease manifests several hours to days after birth, with purpura fulminans (see this term) or massive venous thrombosis. Purpura fulminans is a life-threatening condition involving severe clotting throughout the body and causing necrosis of tissues.
Protein S deficiency is a disorder of blood clotting. People with this condition have an increased risk of developing abnormal blood clots. Individuals with mild protein S deficiency are at risk of a type of clot called a deep vein thrombosis (DVT) that occurs in the deep veins of the arms or legs. If a DVT travels through the bloodstream and lodges in the lungs, it can cause a life-threatening clot known as a pulmonary embolism (PE). Other factors can raise the risk of abnormal blood clots in people with mild protein S deficiency. These factors include increasing age, surgery, immobility, or pregnancy.
Other possible associations include corneal defects, congenital pulmonary stenosis , [19] total anomalous pulmonary venous connection [20] deafness [21] and optic atrophy . [22] Related genes [ edit ] A related gene – Rhomboid domain containing 2 ( RHBDD2 ) – appears to be important in breast cancer . [23] A second related gene – rhomboid family 1 ( RHBDF1 ) – appears to be important in head and neck cancer. [24] A third member of this family – RHBDD1 – cleaves Bcl-2-interacting killer ( BIK ) – a proapoptotic member of the B cell lymphoma 2 ( Bcl-2 ) family. [25] These proteins may also have a role in diabetes . [26] Diagnosis [ edit ] Differential diagnosis [ edit ] The differential diagnosis is quite extensive and includes [27] [28] Buschke–Fischer–Brauer disease Curth–Macklin ichthyosis Gamborg Nielsen syndrome Greither disease Haber syndrome Hereditary punctate palmoplantar keratoderma Jadassohn–Lewandowsky syndrome Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenital and sclerosing keratoderma syndrome Meleda disease Mucosa hyperkeratosis syndrome Naegeli–Franceschetti–Jadassohn syndrome Naxos disease Olmsted syndrome Palmoplantar keratoderma and leukokeratosis anogenitalis Pandysautonomia Papillomatosis of Gougerot and Carteaud Papillon–Lefèvre syndrome Punctate porokeratotic keratoderma Richner–Hanhart syndrome Schöpf–Schulz–Passarge syndrome Unna Thost disease Vohwinkel syndrome Wong's dermatomyositis Treatment [ edit ] Systemic retinoids are the drugs used for tylosis. ... Nihon Kyobu Shikkan Gakkai Zasshi 34(1):76–79 ^ Khanna SK, Agnone FA, Leibowitz AI, Raschke RA, Trehan M (1993) Nonfamilial diffuse palmoplantar keratoderma associated with bronchial carcinoma. J Am Acad Dermatol 28(2 Pt 2):295–297 ^ Murata Y, Kumano K, Tani M, Saito N, Kagotani K (1988) Acquired diffuse keratoderma of the palms and soles with bronchial carcinoma: report of a case and review of the literature.
A number sign (#) is used with this entry because of evidence that tylosis with esophageal cancer is caused by heterozygous mutation in the RHBDF2 gene (614404) on chromosome 17q25.1. Description Palmoplantar keratoderma (PPK) is a complex group of hereditary syndromes that have been classified into diffuse, punctate, and focal forms according to the pattern of hyperkeratosis on the palms and soles (Lucker et al., 1994). For a discussion of phenotypic and genetic heterogeneity of palmoplantar keratoderma, see epidermolytic PPK (144200). Clinical Features Palmoplantar keratoderma (tylosis) was associated with esophageal cancer in 2 kindreds (which perhaps were related) studied in Liverpool by Howel-Evans et al. (1958); the association was first reported by Clarke and McConnell (1954). The disorder is apparently distinct. From Oxford, Shine and Allison (1966) described a 4-generation family cosegregating tylosis and esophageal stricture.
Tylosis with esophageal cancer (TOC) is an inherited condition that increases the risk for esophageal cancer. The symptoms of TOC include thickening of the skin on the palms and soles of the feet (palmoplantar keratoderma) and white lesions inside the mouth. People with TOC are at very high risk to develop esophageal cancer. The palmoplantar keratoderma usually occurs in childhood, and esophageal cancer usually occurs in adulthood. TOC is caused by a variant in the RHBDF2 gene and is inherited in an autosomal dominant pattern. Diagnosis is based on the symptoms, clinical exam, and family history.
A rare genetic disease characterized by thickening of the skin on palms and soles restricted to areas of weight bearing and/or friction (focal, non-epidermolytic palmoplantar keratoderma) and oral and esophageal leukokeratosis, associated with a very high lifetime risk of developing squamous cell carcinoma of the esophagus. The skin lesions appear in childhood and can be complicated by fissuring and infection.
United Cerebral Palsy of the Golden Gate - UCPGG . Retrieved 28 February 2017 . ^ Miller, Freeman; Steven J. ... "The epidemiology of cerebral palsy: incidence, impairments and risk factors". Disabil Rehabil . 28 (4): 183–91. doi : 10.1080/09638280500158422 .
Active or passive exposure to tobacco smoke is suspected of favouring histamine intolerance, but has not been adequately studied. [8] Potentially harmful foods [ edit ] The following food categories have been quoted in literature [9] as histamine rich: Meat and fish [ edit ] Fish products, especially canned fish Ham Offal Pork Salami Smoked meat Other seafood Dairy [ edit ] Matured ("hard") cheeses - the higher degree of ripeness, the higher histamine content Alcohol [ edit ] Beer (especially top-fermented and cloudy/colored) Some French Champagne (made partially with red grapes) Red Wine Fruits, vegetables, legumes and roots [ edit ] Avocado Bamboo sprouts Bananas Beans Citrus fruits Eggplant Figs Garlic Horseradish Mushrooms Papayas Plums Raisins Sauerkraut Spinach Strawberries Tomatoes Other molds (e.g. noble-mold from cheeses and salamis) Other [ edit ] Chocolate (chocolate itself does not contain histamine, but the other biogenic amines from the cocoa do) Nuts [10] Products with vinegar, such as pickles or mustard Soy and soy products (e.g. tofu) (This list is drawn from the German Wikipedia article on histamine intolerance. [11] [ circular reference ] It has been further expanded using Verträglichkeit von histaminhaltigen Lebensmitteln (PDF; 28 kB)). [ specify ] Drug interactions [ edit ] Some medicines [ which? ... The reference ranges (normal values) for blood glutamic acid are 20-107 in infants, 18-65 in children and 28-92 μmol / ml in adults. [15] Literature [ edit ] Abbot, Lieners, Mayer, Missbichler, Pfisterer, Schmutz: Nahrungsmittelunverträglichkeit (Histaminintoleranz) .
Archived from the original on 2019-04-08 . Retrieved 2019-11-28 . ^ a b "OMIM Entry - # 613884 - CHROMOSOME 13q14 DELETION SYNDROME" . www.omim.org . Retrieved 2019-10-08 . ^ "Medical Definition Of Long Arm Of A Chromosome" . www.medicinenet.com/script/main/hp.asp . Retrieved 2019-11-28 . ^ Manolakos, E.; Peitsidis, P.; Garas, A.; Vetro, A.; Eleftheriades, M.; Petersen, M.
Partial monosomy 13q Chromosome 13 which is involved in this condition Partial monosomy of chromosome 13q is a monosomy that results from the loss of all or part of the long arm of chromosome 13 in human beings. It is a rare genetic disorder which results in severe congenital abnormalities which are frequently fatal at an early age. Up until 2003, more than 125 cases had been documented in medical literature. [1] Contents 1 Symptoms 2 Diagnosis 3 Treatment 4 References Symptoms [ edit ] Symptoms vary from case to case, and may correlate to how much of the chromosome is missing. Symptoms that are frequently observed with the condition include: Low birth weight Malformations of the head Eye abnormalities Defects of the hands and feet, polydactyly Reproductive abnormalities (males) Psychological and motor retardation Diagnosis [ edit ] This section is empty. You can help by adding to it . ( December 2017 ) Treatment [ edit ] This section is empty.
"Vulnerable Plaque: Definition, Diagnosis, and Treatment". Cardiology Clinics . 28 (1): 1–30. doi : 10.1016/j.ccl.2009.09.008 . PMID 19962047 . ^ a b "Vulnerable Plaque - Texas Heart Institute Heart Information Center" . www.texasheartinstitute.org . Retrieved 2017-03-28 . ^ a b Robbins and Cotran pathologic basis of disease .
"Old Bone Collection Reveals Basis for Some Dizziness" . The Johns Hopkins Gazette . 28 (25). ^ Duffy, Jim (1999). "The Clue in the Old Bones" . ... External links [ edit ] Ward, Bryan K.; Carey, John P.; Minor, Lloyd B. (28 April 2017). "Superior Canal Dehiscence Syndrome: Lessons from the First 20 Years" .
Superior semicircular canal dehiscence syndrome is a rare balance disorder characterized by auditory and/or vestibular symptoms. These might include dizziness and vertigo triggered by heavy lifting, straining, coughing or loud sounds that change the middle ear or intracranial pressure, fullness in the ear, autophony (an echo or reverberation in the ear when speaking, chewing or swallowing), hearing loss, nystagmus, or oscillopsia (the apparent motion of objects that are stationary). This condition is caused by an opening (dehiscence) in the bone that overlays the superior (uppermost) semicircular canal within the inner ear. While many patients with superior semicircular canal dehiscence syndrome are able to tolerate their symptoms and reduce or avoid triggering stimuli, others can benefit from surgical repair of the dehiscence.
Archived from the original on November 26, 2011 . Retrieved October 28, 2011 . ^ a b EL-Sobky, TA; Shawky, RM; Sakr, HM; Elsayed, SM; Elsayed, NS; Ragheb, SG; Gamal, R (15 November 2017). ... "Blount's Disease" . Retrieved October 28, 2011 . External links [ edit ] "Bowed Legs" .
Mol. Phylogenet. Evol . 47 (2): 717–28. doi : 10.1016/j.ympev.2008.02.015 . ... American Museum of Natural History. 30 July 2008 . Retrieved 28 July 2019 . ^ Auguste AJ, Pybus OG, Carrington CV (2009).
An acute arboviral infection caused by a virus of the Flaviviridae family transmitted by an infected mosquito, and characterized by the onset of flulike symptoms such as fever, malaise, headache, cough, and sore throat that can progress to meningitis or encephalitis with symptoms like nausea, vomiting, confusion, stiff neck, disorientation, irritability, tremors, and convulsions. Photophobia, cranial nerve palsies, and even coma may occur.
Retrieved 2020-04-09 . ^ Grohol, J. (February 28, 2015). "FOMO Addiction: The Fear of Missing Out" . ... Journal of Adolescence, 51, pp. 41-49" (PDF) . University of Glasgow . Retrieved 28 May 2020 . ^ Amichai-Hamburger, Y. & Ben-Artzi, E. (2003), "Loneliness and internet use", Computers in Human Behavior , 19 (1): 71–80, doi : 10.1016/S0747-5632(02)00014-6 ^ Deci, E.L. & Ryan, R.M. (1985).
"Twin Anemia–Polycythemia Sequence in Two Monochorionic Twin Pairs Without Oligo-Polyhydramnios Sequence". Placenta . 28 (1): 47–51. doi : 10.1016/j.placenta.2006.01.010 . ... "Assessment of Feto-fetal Transfusion Flow Through Placental Arterio-venous Anastomoses in a Unique Case of Twin-to-Twin Transfusion Syndrome". Placenta . 28 (2–3): 209–211. doi : 10.1016/j.placenta.2006.03.006 .
Grey et al. (1996) provided a 45-year follow-up on a patient with Engelmann disease initially described by Stronge and McDowell (1950) when he was 28 years of age. The disease had shown progression over the subsequent 45 years, characterized by the unique involvement of the femoral capital epiphyses. ... The R218C mutation appeared to be the most prevalent worldwide, having been found in 17 of 28 reported families. Heterogeneity Campos-Xavier et al. (2001) found no obvious correlation between the nature of TGFB1 mutations and the severity of the clinical manifestations of CED, but observed a marked intrafamilial clinical variability, supporting incomplete penetrance of CED.
Camurati-Engelmann disease is a skeletal condition that is characterized by abnormally thick bones (hyperostosis) in the arms, legs, and skull. The thick limb bones can lead to bone pain and muscle weakness in the arms and legs and cause individuals with Camurati-Engelmann disease to tire quickly. Bone pain ranges from mild to severe and can increase with stress, activity, or cold weather. Leg weakness can make it difficult to stand up from a seated position and some affected individuals develop a waddling or unsteady walk. Additional limb abnormalities include joint deformities (contractures), knock knees, and flat feet (pes planus ).
Camurati-Englemann disease (CED) is a rare, clinically variable bone dysplasia syndrome characterized by hyperostosis of the long bones, skull, spine and pelvis, associated with severe pain in the extremities, a wide-based waddling gait, joint contractures, muscle weakness and easy fatigability. Camurati-Englemann disease (CED) is a rare, clinically variable bone dysplasia syndrome characterized by hyperostosis of the long bones, skull, spine and pelvis, associated with severe pain in the extremities, a wide-based waddling gait, joint contractures, muscle weakness and easy fatigability. Epidemiology The prevalence is unknown but more than 300 cases have been reported to date. CED has been described in various ethnic groups, and males and females are affected equally. Clinical description Most of the clinical signs are related to hyperostosis and sclerosis.
Rare skeletal genetic disorder "Engelmann syndrome" redirects here. For the neuro-genetic disorder, see Angelman syndrome . Camurati–Engelmann disease Other names Engelmann disease (ED) , Engelmann syndrome (ES) , Camurati–Engelmann syndrome (CES) or Progressive diaphyseal dysplasia (PDD) , [1] Osteopathia hyperostotica scleroticans and Multiplex infantalis . [2] This condition is inherited via autosomal dominance Specialty Medical genetics Camurati–Engelmann disease (CED) is a very rare autosomal dominant genetic disorder that causes characteristic anomalies in the skeleton . It is also known as progressive diaphyseal dysplasia. It is a form of dysplasia . [3] Patients typically have heavily thickened bones, especially along the shafts of the long bones (called diaphyseal dysplasia). The skull bones may be thickened so that the passages through the skull that carry nerves and blood vessels become narrowed, possibly leading to sensory deficits, blindness , or deafness . This disease often appears in childhood and is considered to be inherited, however many patients have no previous history of CED within their family.
Summary Clinical characteristics. Camurati-Engelmann disease (CED) is characterized by hyperostosis of the long bones and the skull, proximal muscle weakness, limb pain, a wide-based, waddling gait, and joint contractures. Facial features such as macrocephaly, frontal bossing, enlargement of the mandible, proptosis, and cranial nerve impingement resulting in facial palsy are seen in severely affected individuals later in life. Diagnosis/testing. The diagnosis of CED is established in a proband with the characteristic radiographic findings or (if radiographic findings are inconclusive) on identification of a heterozygous pathogenic variant in TGFB1 by molecular genetic testing. Management. Treatment of manifestations: Corticosteroid therapy as needed to control symptoms; losartan may be a helpful adjuvant therapy to minimize the need for steroids to control pain. Pain is also managed with analgesics and non-pharmacologic methods. Craniectomy may be needed to reduce intracranial pressure and relieve symptoms in individuals with several cranial sclerosis.
Camurati-Engelmann disease is a genetic condition that mainly affects the bones. People with this disease have increased bone density, particularly affecting the long bones of the arms and legs. In some cases, the skull and hip bones are also affected. The thickened bones can lead to pain in the arms and legs, a waddling walk, muscle weakness, and extreme tiredness. The age that symptoms begin varies greatly, but most people with this condition develop pain or weakness by adolescence. Camurati-Engelmann disease is caused by a mutation in the TGFB1 gene and inheritance is autosomal dominant.