Because medication-based non-invasive methods of abortion now exist, dilation and curettage has been declining as a method of abortion, although suction curettage is still the most common method used for termination of a first trimester pregnancy. [6] [7] The World Health Organization recommends D&C with a sharp curette as a method of surgical abortion only when manual vacuum aspiration with a suction curette is unavailable. [8] For patients who have recently given birth, a D&C may be indicated to remove retained placental tissue that does not pass spontaneously or for postpartum hemorrhage. [9] Non-pregnant patients [ edit ] D&Cs for non-pregnant patients are commonly performed for the diagnosis of gynecological conditions leading to abnormal uterine bleeding; [10] to remove the excess uterine lining in women who have conditions such as polycystic ovary syndrome ; [11] to remove tissue in the uterus that may be causing abnormal uterine bleeding, such as endometrial polyps or uterine fibroids ; [3] [2] or to diagnose the cause of post-menopausal bleeding , such as in the case of endometrial cancer . ... Another risk is intrauterine adhesions, or Asherman's syndrome . One study found that in women who had one or two sharp curettage procedures for miscarriage, 14-16% developed some adhesions. [17] Women who underwent three sharp curettage procedures for miscarriage had a 32% risk of developing adhesions. [17] The risk of Asherman's syndrome was found to be 30.9% in women who had D&C following a missed miscarriage, [18] and 25% in those who had a D&C 1–4 weeks postpartum. [19] [20] [21] Untreated Asherman's syndrome, especially if severe, also increases the risk of complications in future pregnancies, such as ectopic pregnancy , miscarriage , and abnormal placentation (e.g. placenta previa and placenta accreta ).
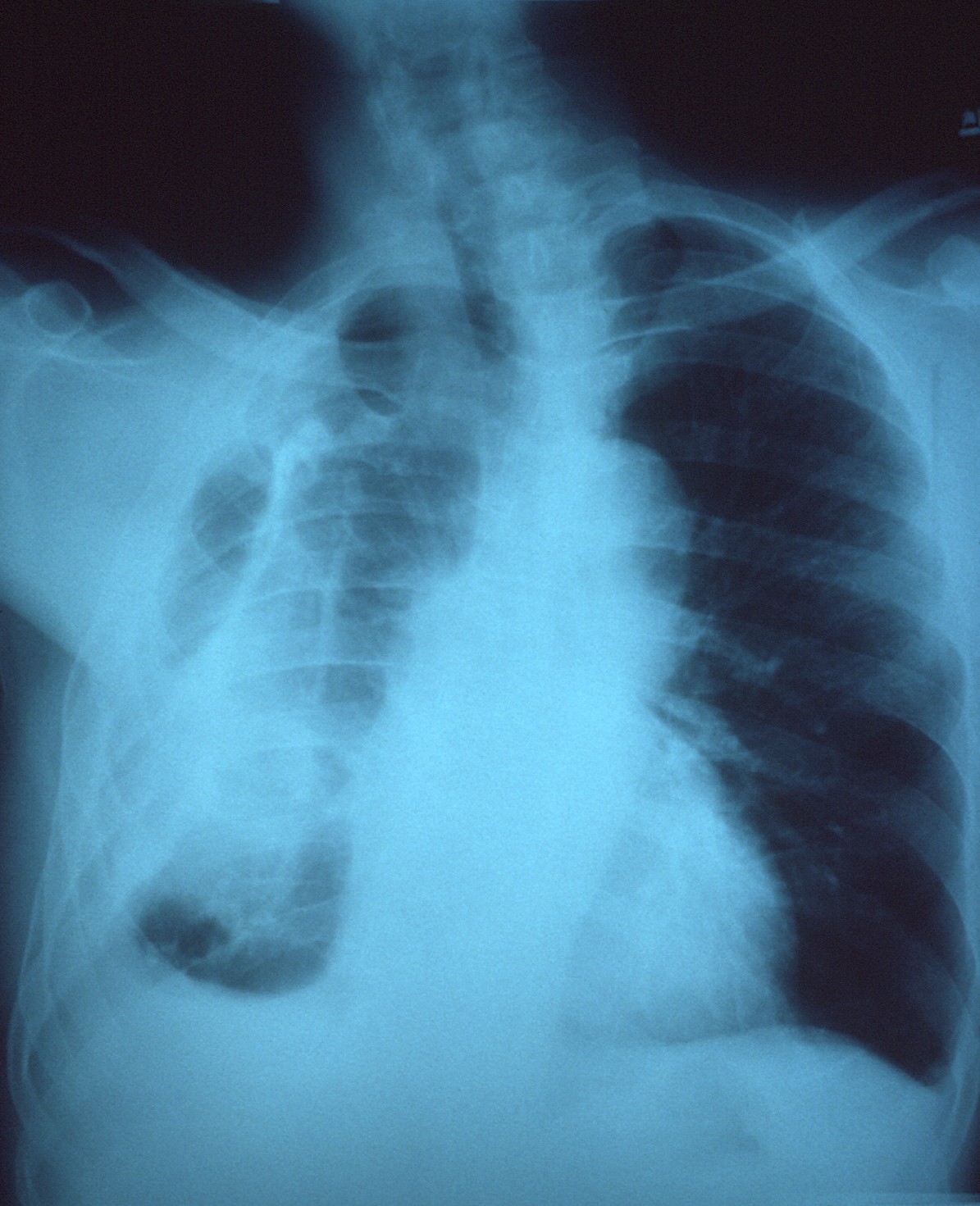
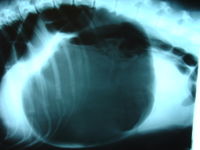
"Canine gastric dilatation/volvulus syndrome in a veterinary critical care unit: 295 cases (1986-1992)". ... "Multiple risk factors for the gastric dilatation-volvulus syndrome in dogs: a practitioner/owner case-control study". ... "A prospective study of survival and recurrence following the acute gastric dilatation-volvulus syndrome in 136 dogs". Journal of the American Animal Hospital Association . 34 (3): 253–9. doi : 10.5326/15473317-34-3-253 . ... "A retrospective study of factors influencing survival following surgery for gastric dilatation-volvulus syndrome in 306 dogs". Journal of the American Animal Hospital Association . 46 (2): 97–102. doi : 10.5326/0460097 .
The flushing that occurs in medullary thyroid carcinoma is indistinguishable from that associated with carcinoid syndrome. In MTC, the flushing, diarrhea, and itching (pruritus) are all caused by elevated levels of calcitonin gene products (calcitonin or calcitonin gene-related peptide ). [4] By comparison, the flushing and diarrhea observed in carcinoid syndrome is caused by elevated levels of circulating serotonin. ... A plasma level of metanephrines should be checked before surgical thyroidectomy takes place to evaluate for the presence of pheochromocytoma since 25% of people found to have medullary thyroid cancer have the inherited form from the MEN2A syndrome. Undiagnosed pheochromocytoma leads to a very high intraoperative risk of hypertensive crisis and, potentially, death. ... "Medullary thyroid carcinoma: including MEN 2A and MEN 2B syndromes". J Surg Oncol . 89 (3): 122–9. doi : 10.1002/jso.20184 .
Medullary thyroid carcinoma (MTC) is developed from thyroid C cells that secrete calcitonin (CT). Epidemiology MTC represents 5-10% of thyroid cancers with a 1-2% incidence in nodular thyroid diseases. Estimated prevalence in the general population is 1/14,300. Diagnostic methods Diagnosis is usually made in the presence of a solitary nodule often associated with nodal metastasis and confirmed by a high basal CT level which represents its biological marker. MTC may present as a sporadic form, and in about 30% of cases, as a familial form as a part of multiple endocrine neoplasia (see this term); a dominant inherited disease related to germline mutation of the protooncogene RET . Both biological (CT) and genetic ( RET ) markers allows optimal diagnosis and treatment of MTC; the former allowing screening and early diagnosis of MTC by routine CT measurements in nodular thyroid diseases that require adequate and complete surgery required to be performed.
Nomenclature Abetalipoproteinemia was initially named Bassen-Kornzweig syndrome. Prevalence Abetalipoproteinemia is rare; fewer than 100 individuals have been described in the literature. ... The only distinguishing feature is the pattern of inheritance: in abetalipoproteinemia the lipid levels in obligate heterozygote parents are normal; in hypobetalipoproteinemia LDL-cholesterol levels are <50% of normal. McLeod neuroacanthocytosis syndrome (MLS) XK XL Acanthocytosis Peripheral neuropathy MLS is X-linked; affected persons have normal lipid profiles & no manifestations of fat-soluble vitamin deficiency (e.g., retinal disease, bone abnormalities, coagulopathy) Friedreich ataxia FXN AR Broad based, high stepping gait Loss of proprioception Loss of deep tendon reflexes Affected persons have normal lipid profiles & no manifestations of fat-soluble vitamin deficiency (e.g., retinal disease, bone abnormalities, coagulopathy) AD = autosomal dominant; AR = autosomal recessive; MOI = mode of inheritance; XL = X-linked Management Evaluations Following Initial Diagnosis To establish the extent of disease and needs in an individual diagnosed with abetalipoproteinemia, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Clinical Features Features are celiac syndrome, pigmentary degeneration of the retina, progressive ataxic neuropathy, and a peculiar 'burr-cell' malformation of the red cells called acanthocytosis.
Abetalipoproteinemia is a very rare condition that affects fat and vitamin absorption by the intestines and liver, leading to very low LDL-cholesterol and malnutrition. Early symptoms of this condition include diarrhea, vomiting, and poor growth. Without treatment, later complications may include muscle weakness, poor night and color vision, tremors, and speech difficulties. The long-term outcome can be difficult to predict. Abetalipoproteinemia is diagnosed based on clinical exam, laboratory tests showing abnormally low cholesterol, and confirmed by genetic testing. This condition is caused by genetic variants in the MTTP gene and is inherited in an autosomal recessive pattern.
Abetalipoproteinemia is an inherited disorder that impairs the normal absorption of fats and certain vitamins from the diet. Many of the signs and symptoms of abetalipoproteinemia result from a severe shortage (deficiency) of fat-soluble vitamins (vitamins A, E, and K). The signs and symptoms of this condition primarily affect the gastrointestinal system , eyes, nervous system, and blood. The first signs and symptoms of abetalipoproteinemia appear in infancy. They often include failure to gain weight and grow at the expected rate (failure to thrive); diarrhea; and fatty, foul-smelling stools (steatorrhea).
A severe, familial hypobetalipoproteinemia characterized by permanently low levels (below the 5th percentile) of apolipoprotein B and LDL cholesterol, and by growth delay, malabsorption, hepatomegaly, and neurological and neuromuscular manifestations. Epidemiology It is very rare, with an estimated prevalence of less than 1/1,000,000. Clinical description Abetalipoproteinemia manifests during the first year of life or in young childhood. It is often associated with growth delay, hepatomegaly with steatosis, diarrhea with steatorrhea, and fat malabsorption. Spastic ataxia, atypical retinitis pigmentosa, acanthocytosis, a low level of liposoluble vitamins, and major cytolysis and even cirrhosis can occur.
11q13.3 Autosomal recessive Slowly progressive Distal spinal muscular atrophy type 4 (DSMA4) 611067 PLEKHG5 1p36.31 Autosomal recessive Slowly progressive, described only in one family Distal spinal muscular atrophy type 5 (DSMA5) 614881 DNAJB2 2q35 Autosomal recessive Young adult onset, slowly progressive Distal spinal muscular atrophy type VA (DSMAVA) Distal hereditary motor neuronopathy type 5A (DHMN5A) 600794 GARS 7p14.3 Autosomal dominant With upper limb predominance; allelic and overlapping with CMT2D , phenotype overlapping with Silver syndrome Distal spinal muscular atrophy type VB (DSMAVB) Distal hereditary motor neuronopathy type 5B (DHMN5B) 614751 REEP1 2p11 Autosomal dominant With upper limb predominance; allelic and overlapping with HSP -31 Distal spinal muscular atrophy with calf predominance Distal hereditary motor neuronopathy type 2D (DHMN2D) 615575 FBXO38 5q32 Autosomal dominant Juvenile- or adult-onset, slowly progressive, affects both proximal and distal muscles, initially manifests with calf weakness which progresses to hands Distal spinal muscular atrophy with vocal cord paralysis Distal hereditary motor neuronopathy type 7A (DHMN7A) Harper–Young myopathy 158580 SLC5A7 2q12.3 Autosomal dominant Adult-onset with vocal cord paralysis, very rare Congenital distal spinal muscular atrophy Distal hereditary motor neuronopathy type 8 (DHMN8) 600175 TRPV4 12q24.11 Autosomal dominant Affects primarily distal muscles of lower limbs, non-progressive, rare, allelic with SPSMA and CMT2C Scapuloperoneal spinal muscular atrophy (SPSMA) Scapuloperoneal neurogenic amyotrophy 181405 TRPV4 12q24.11 Autosomal dominant or X-linked dominant Affects muscles of lower limbs, non-progressive, rare, allelic with congenital distal spinal muscular atrophy and CMT2C Autosomal dominant distal spinal muscular atrophy Distal hereditary motor neuronopathy type 2A (DHMN2A) 158590 HSPB8 12q24.23 Autosomal dominant Adult-onset. ... "The spectrum of lower motor neuron syndromes" . Journal of Neurology . 250 (11): 1279–92. doi : 10.1007/s00415-003-0235-9 . ... External links [ edit ] Classification D ICD - 10 : G12 MeSH : D009134 v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
Disorder where there is enlargement of both legs due to deposits of fat under the skin Lipedema Other names Lipoedema, lipödem, lipalgia, adiposalgia, adipoalgesia, adiposis dolorosa, lipomatosis dolorosa of the legs, lipohypertrophy dolorosa, painful column leg, painful lipedema syndrome A very advanced case of lipedema of the right leg (the knee is pointing to the right and is concealed by the overhanging lipedema). ... Many clinicians are unaware of the disease. [4] Trayes 2013 published some tools including tables and a flow chart that can be used to diagnose lipedema and other edemas. [9] Lipo-lymphedema [ edit ] Lipo-lymphedema, a secondary lymphedema, is associated with both lipedema and obesity (which occur together in the majority of cases), most often lipedema stages 2 and 3. [12] Dercum's disease [ edit ] Lipedema / Dercum's disease differentiation – these conditions may co-exist. Dercum's disease is a syndrome of painful growths in subcutaneous fat. ... "Lipedema: An overview of its clinical manifestations, diagnosis and treatment of the disproportional fatty deposition syndrome - systematic review" . Clinical Obesity . 2 (3–4): 86–95. doi : 10.1111/j.1758-8111.2012.00045.x . ... J Dtsch Dermatol Ges. 2017;15(7):758-767. doi: 710.1111/ddg.13036 ^ a b Wold, LE; Hines, EA; Allen, EV (1 May 1951). "Lipedema of the legs: a syndrome characterized by fat legs and edema".
Child et al. (2010) also identified 4 unrelated isolated adult males with lipedema: 2 were testosterone-depleted, 1 had liver disease, gynecomastia, and signs of estrogen excess, and 1 had growth hormone deficiency and hypogonadism. None had syndromic features, and none had a family history of lipedema.
Significant genetic correlations were observed between HMW adiponectin and fasting insulin, homeostasis models of insulin resistance, HDL cholesterol, and metabolic syndrome score. The SNP rs1501299, previously reported as 276G-T, was found to account for the correlation between HMW adiponectin and insulin and metabolic syndrome score.
Unger et al. (2008) reported 6 additional patients with Goldblatt syndrome, including a second sib pair (brother and sister). ... Exclusion Studies Unger et al. (2008) performed complete sequencing of several genes, including the COL2A1 gene, in one or more of the patients they reported with Goldblatt syndrome and identified no pathogenic mutations.
Clinical Features Al-Gazali et al. (2001) reported 5 children from 2 consanguineous families, of Palestinian and Syrian origin, respectively, with features overlapping both geroderma osteodysplastica (GO; 231070) and wrinkly skin syndrome (WSS; 278250). All 5 children had similar dysmorphic facial features, consisting of broad and prominent forehead, hypotelorism with epicanthal folds, prominent bulbous nose, flat malar region, and large protruding ears. ... In patients with autosomal recessive cutis laxa, including those previously described by Al-Gazali et al. (2001), Hamamy et al. (2005), Nanda et al. (2008), and Rajab et al. (2008) with clinical features of geroderma osteodysplastica and/or wrinkly skin syndrome, Reversade et al. (2009) sequenced the PYCR1 gene and identified homozygosity or compound heterozygosity for 6 missense mutations, 1 frameshift mutation, 5 splice site mutations, and a 22-bp deletion (see, e.g., 179035.0002-179035.0008).
A rare, hereditary, developmental defect with connective tissue involvement characterized by cutis laxa of variable severity, in utero growth restriction, congenital hip dislocation and joint hyperlaxity, wrinkling of the skin, in particular the dorsum of hands and feet, and progeroid facial features. Hypotonia, developmental delay, and intellectual disability are common. In addition, cataracts, corneal clouding, wormian bones, lipodystrophy and osteopenia have been reported.
Thus, the gene for this disorder, designated ALS4, is genetically distinct from previously mapped familial ALS syndromes. Blair et al. (2000) refined the position of the ALS4 locus to a critical interval of less than 3 cM on 9q34. In 3 families with a clinical syndrome with similarities to both ALS4 and dHMN, De Jonghe et al. (2002) found positive linkage (lod scores greater than 3) with markers located within the ALS4 locus region on 9q34.
A rare, genetic motor neuron disease characterized by late childhood- or adolescent-onset of slowly progressive, severe, distal limb muscle weakness and wasting, in association with pyramidal signs, normal sensation, and absence of bulbar involvement, leading to degeneration of motor neurons in the brain and spinal cord.
Description Congenital disorders of glycosylation, previously called carbohydrate-deficient glycoprotein syndromes (CDGSs), are caused by defects in mannose addition during N-linked oligosaccharide assembly. ... At 18 years, she had severe premenstrual syndrome, but menses were regular. She had normal secondary sex characteristics.
ALG6 -congenital disorder of glycosylation ( ALG6 -CDG, also known as congenital disorder of glycosylation type Ic) is an inherited condition that affects many parts of the body. The signs and symptoms of ALG6 -CDG vary widely among people with the condition. Individuals with ALG6 -CDG typically develop signs and symptoms of the condition during infancy. They may have difficulty gaining weight and growing at the expected rate (failure to thrive). Affected infants often have weak muscle tone (hypotonia) and developmental delay.
A form of congenital disorders of N-linked glycosylation characterized by feeding problems, mild-to-moderate neurologic involvement with hypotonia, poor head control, developmental delay, ataxia, strabismus, and seizures, ranging from febrile convulsions to epilepsy. Retinal degeneration has also been reported. A minority of patients show other manifestations, particularly intestinal (such as protein-losing enteropathy) and liver involvement. The disease is caused by loss of function mutations of the gene ALG6 (1p31.3).
Clinical Features Dupuis et al. (2003) studied 2 unrelated infants, P1 and P2, with a clinical syndrome of severe mycobacterial and viral diseases not consistent with any known primary immunodeficiency. ... Molecular Genetics In 2 unrelated infants, P1 and P2, with a clinical syndrome of severe mycobacterial and viral diseases, Dupuis et al. (2003) identified homozygosity for point mutations in the STAT1 gene (600555.0002; 600555.0003).
Susceptibility to viral and mycobacterial infections is a rare, genetic, primary immunodeficiency due to a defect in innate immunity disorder characterized by impaired intracellular signaling from both type I and type II interferons, leading to early-onset, severe, life-threatening intracellular bacterial (typically mycobacteria) and viral (mainly herpes viruses) infections.
'Autism spectrum disorder,' sometimes referred to as ASD, is a broader phenotype encompassing the less severe disorders Asperger syndrome (see ASPG1; 608638) and pervasive developmental disorder, not otherwise specified (PDD-NOS). ... Mental retardation coexists in approximately two-thirds of individuals with ASD, except for Asperger syndrome, in which mental retardation is conspicuously absent (Jones et al., 2008).
Dermographism is a form of urticaria in which a single stroke or scratch of the skin produces a localized wheal with surrounding red flare. It is the most common form of physical urticaria. Although familial cold urticaria (120100) is well known, the first report of familial dermographism is apparently that by Jedele and Michels (1991). They observed 5 affected individuals in 4 generations with no male-to-male transmission. They illustrated the wheal formation on the skin of the back after a single stroke with moderate pressure. Inheritance - Autosomal dominant Skin - Dermographism - Localized urticarial wheal and flare from a single skin stroke or scratch ▲ Close
Familial dermographism is a condition also known as skin writing. When people who have dermatographia lightly scratch their skin, the scratches redden into a raised wheal similar to hives. Signs and symptoms of dermatographia include raised red lines, swelling, inflammation, hive-like welts and itching. Symptoms usually disappear within 30 minutes. The exact cause of this condition is unknown. Treatment may invovle use of antihistamines if symptoms do not go away on their own.
P. (2007). A Clinical Guide to Epileptic Syndromes and Their Treatment . Springer . pp. 107–112. ... Benign childhood partial seizures and related epileptic syndromes . John Libbey Eurotext. pp. 288–291.
Journal of the History of the Behavioral Sciences : Vol 11, Issue 1: "Agalmatophilia, the statue syndrome." Wiley Periodicals, Inc. Simmons, Laurence (2006). ... Journal of Sex Research ; Vol. 14, Issue 4: "The Statue Syndrome: Perversion? Fantasy? Anecdote?".