-
Cushing's Syndrome
Wikipedia
While all Cushing's disease gives Cushing's syndrome, not all Cushing's syndrome is due to Cushing's disease. Several possible causes of Cushing's syndrome are known. [ citation needed ] Exogenous [ edit ] The most common cause of Cushing's syndrome is the use of prescribed glucocorticoids to treat other diseases ( iatrogenic Cushing's syndrome). ... The ACTH levels remain high because the tumor is unresponsive to negative feedback from high cortisol levels. When Cushing's syndrome is due to extra ACTH it is known as ectopic Cushing syndrome. [41] This may be seen in a paraneoplastic syndrome . ... This clinical situation is known as Nelson's syndrome . [50] Epidemiology [ edit ] Cushing's syndrome caused by treatment with corticosteroids is the most common form. ... PMID 20829610 . ^ "Cushing Syndrome: Condition Information" . 2012-11-30.POMC, NR3C1, HSD11B1, CYP11B1, GH1, VWF, CRH, ARMC5, CYP11B2, IGF1, PRKACA, PRKAR1A, SERPINE1, REN, LEP, PPARG, PLG, SLC6A4, BRD2, KCNJ5, SST, SULT2A1, PHLDA2, TWIST1, CD163, SOST, LOC110673971, KRT5, ACP5, IL10, ACE, BDNF, CETP, CHGA, COX8A, CPA1, CRHR1, CRP, CYP19A1, F12, BAG1, GABRA6, GIP, GIPR, GNAS, HMGCR, NR4A1, HSD3B1, HSD3B2, LOC110673972

-
Emberger Syndrome
Wikipedia
Emberger syndrome This syndrome is autosomal dominant The Emberger syndrome is a rare, autosomal dominant , genetic disorder caused by familial or sporadic inactivating mutations in one of the two parental GATA2 genes. ... Individuals with the Emberger syndrome may exhibit signs or symptoms that are more characteristic of the latter manifestations. ... Other defects less commonly associated with and the syndrome include hypotelorism , epicanthic folds , hydrocele , webbed neck , and warts caused be human papillomavirus infection. [1] [5] [7] In these case of relatively benign symptoms and signs, the syndrome commonly progresses rapidly or slowly to myelodysplastic syndrome followed by acute myeloid leukemia . ... It is not exactly clear how reduced levels of GATA2 cause any of Emberger syndrome's hematological disorders. [6] The role of GATA2 in promoting the normal development of the lymphatic stem cells may be responsible for the other two key features of the Emberger syndrome. ... However, treatment of the disorder's myelodysplastic syndrome and acute myeloid leukemia differs somewhat from standard measures.

-
Laugier–hunziker Syndrome
Wikipedia
Laugier–Hunziker syndrome Specialty Dermatology Laugier–Hunziker syndrome ( / ˈ l oʊ ʒ i eɪ ˈ h ʊ n t s ɪ k ər / ) is a cutaneous condition characterized by hyperpigmentation of the oral mucosa , [1] longitudinal melanonychia , [1] and genital melanosis . [2] The hyperpigmentation presented in Laugier-Hunziker syndrome is benign and should be differentiated from Peutz-Jeghers syndrome. See also [ edit ] Peutz–Jeghers syndrome List of cutaneous conditions References [ edit ] ^ a b Nayak, RS; et al. (2012), "Laugier–Hunziker syndrome", J Oral Maxillofac Pathol , 16 (2): 245–250, doi : 10.4103/0973-029X.99079 , PMC 3424942 , PMID 22923898 . ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007).

-
Cushing Syndrome
Mayo_clinic
Cushing syndrome also can cause high blood pressure or bone loss. ... Causes Cushing syndrome is caused by having too much cortisol in the body. ... The role of glucocorticoid medicines (exogenous Cushing syndrome) Cushing syndrome can happen from taking glucocorticoid medicines. ... Some tumors make ACTH, which creates more cortisol and can cause Cushing syndrome. Problems with the adrenal glands also can affect cortisol and cause Cushing syndrome. ... But they can make cortisol and cause Cushing syndrome. Sometimes, several lumps that make cortisol can grow in the adrenal glands and cause Cushing syndrome.CRH, POMC, PRKAR1A, PRKACB, NR3C1, USP8, CDH23, GABRA6, DRD2, CYBB, SERPINA1, GIPR, PRKACA, GIP, ARMC5, CYP11B1, PDE11A, VWF, HSD11B1, MEN1, CYP17A1, GNAS, SST, CGB3, CGA, HSD11B2, HTC2, REN, CGB5, CGB8, PDE8B, CTNNB1, MC2R, GH1, GHRL, SULT2A1, IL6, FDXR, RNU1-4, PRL, LEP, LHCGR, TP53, NR3C2, AGT, DST, TXNIP, AKR1B1, RNU1-1, BDNF, PHLDA2, LGR6, NDRG2, GBA3, SLC25A19, PPARG, BRD2, POR, COASY, FBXO32, RET, BCOR, TWIST1, SLC8A1, KRT20, SOST, SLC12A3, SMUG1, PMPCA, UTS2, C1QL1, HDAC9, BEST1, STAR, TXN, CCDC6, SERPINA3, PECAM1, TSC22D3, ACE, CYP11B2, CYP3A4, CYB5A, CPA1, COX8A, CNC2, CETP, SCARB1, SERPINA6, CAMP, CACNA1D, BGLAP, AVP, ATR, APRT, AGTR2, AGTR1, AGRP, ADRA2A, ACP5, DMD, E2F1, MFAP1, F2, CYP4F3, KRT5, KCNJ5, IRF6, IL18, CXCL8, ACAT1, IGF2, IGF1R, IGF1, HTR4, GSTA1, GHSR, FOLH1, FOXO3, FKBP5, FH, FDX1, FASN, F12, F5, PGR-AS1
-
Yellow Nail Syndrome
Omim
Description Yellow nail syndrome (YNS) is classically considered to comprise a clinical triad of yellow nails, lymphedema, and respiratory tract involvement. ... Finegold et al. (2001) concluded that there is phenotypic overlap between lymphedema-distichiasis syndrome and lymphedema-yellow nail syndrome. ... Rezaie et al. (2008) suggested that lymphedema with yellow nails is not a distinct disorder but rather in the spectrum of lymphedema-distichiasis syndrome. They noted that 6 of the 7 patients in the family of Finegold et al. (2001) reported as having yellow-nail syndrome also had distichiasis, which is characteristic of the lymphedema-distichiasis syndrome. ... In yellow nail syndrome, the nail plate is yellow and overcurved, but remains translucent and smooth. By contrast, yellow nails in lymphedema syndromes become thickened, rough, and opaque.
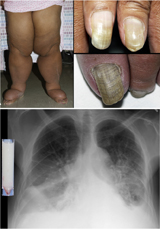
-
Hermansky-Pudlak Syndrome
Medlineplus
People with Hermansky-Pudlak syndrome also have problems with blood clotting (coagulation ) that lead to easy bruising and prolonged bleeding. ... Other, less common features of Hermansky-Pudlak syndrome include inflammation of the large intestine (granulomatous colitis) and kidney failure. There are nine different types of Hermansky-Pudlak syndrome, which can be distinguished by their signs and symptoms and underlying genetic cause. ... The other genes associated with Hermansky-Pudlak syndrome each account for a small percentage of cases of this condition. In some people with Hermansky-Pudlak syndrome, the genetic cause of the disorder is unknown.
-
Bardet-Biedl Syndrome
Medlineplus
Bardet-Biedl syndrome is a disorder that affects many parts of the body. ... Frequency In most of North America and Europe, Bardet-Biedl syndrome has a prevalence of 1 in 140,000 to 1 in 160,000 newborns. ... Researchers believe that defective cilia are responsible for most of the features of Bardet-Biedl syndrome. About one-quarter of all cases of Bardet-Biedl syndrome result from mutations in the BBS1 gene. ... In about 25 percent of people with Bardet-Biedl syndrome, the cause of the disorder is unknown. ... Learn more about the genes associated with Bardet-Biedl syndrome BBS1 BBS10 CEP290 MKKS Additional Information from NCBI Gene: ARL6 BBS12 BBS2 BBS4 BBS5 BBS7 BBS9 MKS1 TRIM32 TTC8 Inheritance Pattern Bardet-Biedl syndrome is typically inherited in an autosomal recessive pattern , which means both copies of a BBS gene in each cell have mutations.
-
Hennekam Syndrome
Medlineplus
Hennekam syndrome is an inherited disorder resulting from malformation of the lymphatic system, which is part of both the circulatory system and immune system. ... The signs and symptoms of Hennekam syndrome vary widely among affected individuals, even those within the same family. ... Frequency At least 50 cases of Hennekam syndrome have been reported worldwide. Causes Mutations in the CCBE1 or FAT4 gene can cause Hennekam syndrome. ... CCBE1 gene mutations that cause Hennekam syndrome change the three-dimensional shape of the protein and severely decrease its function. ... FAT4 gene mutations that cause Hennekam syndrome result in a FAT4 protein with decreased function.
-
Anticonvulsant Hypersensitivity Syndrome
Wikipedia
Anticonvulsant hypersensitivity syndrome Anticonvulsant/sulfonamide hypersensitivity syndrome is a potentially serious hypersensitivity reaction that can be seen with medications with an aromatic amine chemical structure, such as aromatic anticonvulsants (e.g. diphenylhydantoin , phenobarbital , phenytoin , carbamazepine , lamotrigine ), sulfonamides , or other medications with an aromatic amine (e.g., procainamide ). Cross-reactivity should not occur between medications with an aromatic amine and medications without an aromatic amine (e.g., sulfonylureas , thiazide diuretics , furosemide , and acetazolamide ); therefore, these medications can be safely used in the future. [1] The hypersensitivity syndrome is characterized by a rash that is initially rash that appears similar to measles (morbilliform) . [2] : 118 The rash may also be one of the potentially lethal severe cutaneous adverse reactions , the DRESS syndrome , Stevens–Johnson syndrome , or toxic epidermal necrolysis . [3] [4] Systemic manifestations occur at the time of skin manifestations and include a high number of eosinophils in the blood , liver inflammation , and interstitial nephritis . ... The risk of first-degree relatives developing the same hypersensitivity reaction is higher than in the general population. [1] As this syndrome can present secondary to multiple anticonvulsants, the general term "anticonvulsant hypersensitivity syndrome" (AHS) is favored over the original descriptive term "dilantin hypersensitivity syndrome." [2] : 118 As of 2015, two cases of AHS have been reported that manifested during long-term treatment with multiple anti-seizure medications . ... "Current Perspectives on Stevens–Johnson Syndrome and Toxic Epidermal Necrolysis". ... "Late-onset Anticonvulsant Hypersensitivity Syndrome Mimicking Lymphoma" . Internal Medicine . 54 (24): 3201–3204. doi : 10.2169/internalmedicine.54.5111 .
-
Wallis–zieff–goldblatt Syndrome
Wikipedia
Wallis–Zieff–Goldblatt syndrome Other names Cleidorhizomelic syndrome Wallis–Zieff–Goldblatt syndrome has an autosomal dominant pattern of inheritance. Wallis–Zieff–Goldblatt syndrome is a rare condition characterized by inherited skeletal disorders manifested mainly as rhizomelic short stature and lateral clavicular defects. [1] It is also known as Cleidorhizomelic syndrome . [2] Contents 1 Presentation 2 References 3 External links Presentation [ edit ] An initial clinical report of this syndrome describes a 6-month-old boy with rhizomelic shortening, particularly in the arms, and protuberances over the lateral aspects of the clavicles . ... "Newly recognized autosomal dominant syndrome of rhizomelic shortness with clavicular defect". ... PMID 3239579 . ^ Online Mendelian Inheritance in Man (OMIM): Cleidorhizomelic syndrome - 119650 External links [ edit ] Classification D ICD - 10 : Q77.8 OMIM : 119650 MeSH : C536428 External resources Orphanet : 1453 v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline This article about a disease of musculoskeletal and connective tissue is a stub .
-
Levator Ani Syndrome
Wikipedia
Levator ani syndrome Other names Levator spasm , Puborectalis syndrome , Chronic proctalgia , Piriformis syndrome , Pelvic tension myalgia , Levator syndrome , and Proctodynia Left levator ani muscle seen from within Specialty Gastroenterology Symptoms Brief intermittent burning anorectal pain or tenesmus Causes Painful spasm of the levator ani muscle Treatment Walking, pelvic relaxation techniques, massage, warm baths, muscle relaxant medications Levator ani syndrome is a condition characterized by brief intermittent burning pain or tenesmus of the rectal or perineal area, [1] caused by spasm of the levator ani muscle. [2] [3] [4] The genesis of the syndrome is unknown; however, inflammation of the arcus tendon is a possible cause of levator ani syndrome. [5] Contents 1 Signs and symptoms 2 Cause 3 Diagnosis 4 Treatment 5 References 6 External links Signs and symptoms [ edit ] Symptoms include a dull ache to the left 2 inches above the anus or higher in the rectum and a feeling of constant rectal pressure or burning. ... Palpation of the levator ani muscle may find tenderness. [7] Cause [ edit ] Levator ani syndrome is characterized by painful spasm of the levator ani muscle. [2] [3] [4] The genesis of the syndrome is unknown, however it has been suggested that inflammation of the arcus tendon is the possible cause of levator ani syndrome. [5] Proctalgia fugax and levator ani syndrome have not been found to be of psychosomatic origin, although stressful events may trigger attacks. [3] Occurrence of levator ani syndrome is associated with "significant elevations on the hypochondriasis, depression, and hysteria scales of the Minnesota Multiphasic Personality Inventory ," which is also the case in general among chronic pain sufferers. [4] Diagnosis [ edit ] The diagnosis of levator ani syndrome is clinical, based on the pattern of signs and symptoms. The diagnosis does not require any routine imaging or additional testing, though other causes of rectal pain must be excluded. Suspected levator ani syndrome is confirmed in the presence of chronic or recurrent rectal pain, occurring in episodes that last at least 30 minutes, with tenderness with posterior traction of the puborectalis muscle. ... ISBN 978-1-4160-2999-1 . ^ a b Levator Syndrome, by Parswa Ansari, MD 7/2014, Merck Manuals ^ a b c Giulio Aniello Santoro; Andrzej Paweł Wieczorek; Clive I. ... "Comparison study between electrogalvanic stimulation and local injection therapy in levator ani syndrome". International Journal of Colorectal Disease . 20 (3): 272–6. doi : 10.1007/s00384-004-0662-9 .

-
Nicolaides-Baraitser Syndrome
Medlineplus
Nicolaides-Baraitser syndrome is a condition that affects many body systems. ... In people with Nicolaides-Baraitser syndrome, the sparse scalp hair is often noticeable in infancy. ... Most people with Nicolaides-Baraitser syndrome have epilepsy, which often begins in infancy. ... Almost everyone with Nicolaides-Baraitser syndrome has moderate to severe intellectual disability. ... Causes Nicolaides-Baraitser syndrome is caused by mutations in the SMARCA2 gene.
-
Schwartz–jampel Syndrome
Wikipedia
Schwartz–Jampel syndrome Other names Myotonic myopathy, dwarfism, chondrodystrophy, ocular and facial anomalies, Dysostosis enchondralis metaepiphysaria, Catel-Hempel type Schwartz–Jampel syndrome is inherited in an autosomal recessive manner. ... Y.; King, Nigel M. (June 2012). "Schwartz-Jampel syndrome: a review of the literature and case report". ... PMID 22591433 . ^ a b c d e f "Schwartz Jampel syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . Retrieved 2018-03-05 . ^ Stum, M; Davoine, CS; Fontaine, B; Nicole, S (Oct 2005). "Schwartz-Jampel syndrome and perlecan deficiency". Acta Myologica . 24 (2): 89–92. PMID 16550923 . ^ Ault, Jennifer (2012-02-01). "Schwartz-Jampel Syndrome" . Medscape . ^ Schwartz, O; Jampel, R.HSPG2, ADAMTSL2, SOST, KDR, VEGFA, GLB1, CHST3, FLT1, FLNA, DLL3, TRAPPC2, BCL2, COL11A1, COL9A1, COL2A1, TRPV4, HLA-C, HLA-B, HLA-A, GNLY, HLA-DRB1, TLR3, HLA-DQB1, RBM45, GZMB, IKZF1, CYP2C9, TNF, CYP2B6, EGFR, CCL17, MIR18A, MIR146A, BAG6, TLR2, TRBV20OR9-2, TNFSF14, MARCHF1, WASF1, SPAG9, PPIG, ABCG2, PANK2, SPTBN2, TRAF3IP2, CCL27, SETX, ACHE, IL4, SGSH, PTGER3, ANXA1, FASLG, C3AR1, C5AR1, TPP1, CRP, CYP2C19, CYP2C18, ATN1, DSP, EPHX1, FDXR, FPR1, GFER, GJA1, GPT, GSTM1, CFH, HMGB1, AKR1B1, IL4R, IL13, ABCB1, SERPINA1, PPARG, MICA

-
47, Xyy Syndrome
Gard
47, XYY syndrome is a syndrome (group of signs and symptoms) that affects males. For some males with this syndrome, signs and symptoms are barely noticeable. For others, signs and symptoms may include learning disabilities , speech delay , low muscle tone (hypotonia), and being taller than expected. 47, XYY syndrome is caused by having an extra copy of the Y chromosome in every cell of the body. The syndrome is usually not inherited. Diagnosis can be made based on prenatal tests, or it may occur during childhood or adulthood if a male has signs or symptoms of the disease.
-
Kallmann Syndrome-Heart Disease Syndrome
Orphanet
Kallmann syndrome with cardiopathy is characterised by hypogonadotropic hypogonadism associated with gonadotropin-releasing hormone (GnRH) deficiency, anosmia or hyposmia (with hypoplasia or aplasia of the olfactory bulbs) and complex congenital cardiac malformations (double-outlet right ventricle, dilated cardiomyopathy, right aortic arch). It represents a distinct clinical entity from Kallmann syndrome. Epidemiology Less than 10 cases have been described so far.
-
Adams-Oliver Syndrome
Medlineplus
Adams-Oliver syndrome is a rare condition that is present at birth. ... Frequency Adams-Oliver syndrome is a rare disorder; its prevalence is unknown. ... The NOTCH1 and DLL4 gene mutations involved in Adams-Oliver syndrome likely impair Notch1 signaling, which may underlie blood vessel and heart abnormalities in some people with Adams-Oliver syndrome. ... These changes in gene activity impair the proper development of the skin, bones, and other tissues, leading to the features of Adams-Oliver syndrome. Little is known about how mutations in the EOGT gene cause Adams-Oliver syndrome. ... Learn more about the genes associated with Adams-Oliver syndrome ARHGAP31 DLL4 DOCK6 EOGT NOTCH1 RBPJ Inheritance Pattern Adams-Oliver syndrome can have different inheritance patterns.
-
Gourmand Syndrome
Wikipedia
Rare eating disorder caused by injury to the frontal lobe or limbic structures Gourmand syndrome Frontal lobe (at right) Specialty Neurology Gourmand syndrome is a very rare and benign eating disorder that usually occurs six to twelve months after an injury to the frontal lobe . [1] [2] [3] [4] Those with the disorder usually have suffered from a right hemisphere frontal or temporal brain lesion typically affecting the cortical areas , basal ganglia or limbic structures. [3] [2] [5] [6] These people develop a new, post-injury passion for gourmet food. [3] [2] [5] [4] There are two main aspects of gourmand syndrome: first, the fine dining habits and changes to taste, and second, the obsessive component, which may result in craving and preservation. [2] Gourmand syndrome can be related to, and shares biological features with, addictive and obsessive disorders. [2] [3] The syndrome was first characterised in 1997. [3] Contents 1 Signs and symptoms 2 Causes 3 History 4 References 5 Further reading Signs and symptoms [ edit ] A new-found obsession for fine foods [2] [4] [3] [6] [5] [1] [ excessive citations ] Wanting to write, talk, eat, about refined foods [2] [1] [3] [4] [5] [6] [ excessive citations ] Causes [ edit ] It is believed that the frontotemporal circuits, normally involved in healthy eating, can, when injured, cause gourmand syndrome in patients. [4] History [ edit ] Only 36 people had been diagnosed with gourmand syndrome as of 2001. [6] In many of these cases, the patient did not have any interest in food beforehand nor had any family history with eating disorders. [5] [2] [3] The first, most famous case was seen in 1997 by Regard and Landis in the journal Neurology : [2] [3] after a Swiss stroke patient was released from the hospital, he immediately quit his job as a political journalist and took up the profession of food critic. [3] Regard and Landis also observed an athletic businessman with this condition whose family was shocked to see such a sudden, drastic change in his diet. [3] Only one case of gourmand syndrome has been reported in a child. ... PMID 17456824 . ^ a b c d e f g h i j Kurian, M.; Schmitt-Mechelke, T.; Korff, C.; Delavelle, J.; Landis, T.; Seeck, M. (2008). " " Gourmand syndrome" in a child with pharmacoresistant epilepsy". ... PMID 18502182 . ^ a b c d e f g h i j k Regard, Marianne; Landis, Theodor (1997). " " Gourmand syndrome": Eating passion associated with right anterior lesions". ... Bramen, Lisa (2011-07-06). "Gourmand Syndrome – First identified by neuroscientists in the 1990s, the disorder is marked by "a preoccupation with food and a preference for fine eating" . ... Retrieved 27 April 2015 . v t e Neurotrauma Traumatic brain injury Intracranial hemorrhage Intra-axial Intraparenchymal hemorrhage Intraventricular hemorrhage Extra-axial Subdural hematoma Epidural hematoma Subarachnoid hemorrhage Brain herniation Cerebral contusion Cerebral laceration Concussion Post-concussion syndrome Second-impact syndrome Dementia pugilistica Chronic traumatic encephalopathy Diffuse axonal injury Abusive head trauma Penetrating head injury Spinal cord injury Anterior spinal artery syndrome Brown-Séquard syndrome Cauda equina syndrome Central cord syndrome Paraplegia Posterior cord syndrome Spinal cord injury without radiographic abnormality Tetraplegia (Quadriplegia) Peripheral nerves Nerve injury Peripheral nerve injury classification Wallerian degeneration Injury of accessory nerve Brachial plexus injury Traumatic neuroma This article about a medical condition affecting the nervous system is a stub .

-
Blue Toe Syndrome
Wikipedia
Blue toe syndrome Specialty Cardiology Blue toe syndrome is a situation that may reflect atherothrombotic microembolism , causing transient focal ischaemia, occasionally with minor apparent tissue loss, but without diffuse forefoot ischemia . [1] The development of blue or violaceous toes can also occur with trauma, cold-induced injury, disorders producing generalized cyanosis, decreased arterial flow, impaired venous outflow, and abnormal circulating blood. [2] [3] [4] [5] [6] [7] [8] [9] The terms "blue toe syndrome", "grey toe syndrome" and "purple toe syndrome" are sometimes used interchangeably. [10] Studies may include echocardiography , thoracic and abdominal CT or MRI, [11] [12] [13] [14] [15] [16] [17] [18] [ excessive citations ] peripheral arterial run off imaging studies, [19] hypercoagulopathy labs, [20] and interrogation of syndromes that lead to peripheral vascular pathology. [21] See also [ edit ] Warfarin#Purple toe syndrome Cholesterol embolism References [ edit ] ^ 'Standards for vascular reporting' ^ Matchett WJ, McFarland DR, Eidt JF, Moursi MM (2000). "Blue toe syndrome: treatment with intra-arterial stents and review of therapies". ... "[Cholesterol (cholestelin) embolization syndrome--blue toe syndrome]". Ryoikibetsu Shokogun Shirizu (14): 469–72. ... External links [ edit ] Pictures are available on this link. 'Blue toe syndrome' Nijhof IS, Majoie IM, Dijkhorst-Oei LT, Bousema MT (2007). "[Blue toe syndrome; a sign of end-arterial occlusion]".
-
Lujan-Fryns Syndrome
Omim
Opitz-Kaveggia syndrome (OKS; 305450) is an allelic disorder with an overlapping phenotype. ... All were referred to the genetics clinic with the diagnosis of Marfan syndrome (154700). Gurrieri and Neri (1991) observed the syndrome in brother and sister. ... Fryns (1993) found 18 cases of this disorder among 682 cases of syndromic mental retardation; the fragile X syndrome (300624), the Aarskog syndrome (305400), and the Coffin-Lowry syndrome (303600) represented 560, 60, and 20 cases, respectively. ... The authors suggested that the similarity of Lujan-Fryns syndrome to Marfan syndrome and the presence of aortic root dilation in their patients may implicate a mutation in a structural connective tissue gene in the etiology of this condition. ... The findings indicated that Lujan-Fryns syndrome and Opitz-Kaveggia syndrome are allelic disorders.

-
3mc Syndrome 2
Omim
Description The term '3MC syndrome' encompasses 4 rare autosomal recessive disorders that were previously designated the Carnevale, Mingarelli, Malpuech, and Michels syndromes, respectively. ... Titomanlio et al. (2005) reviewed the phenotypic similarities between Michels syndrome (257920), Malpuech syndrome (248340), Carnevale syndrome and OSA syndrome, and suggested that they may represent a single recessive spectrum rather than separate disorders. Titomanlio et al. (2005) proposed that the combined entity could be referred to as the '3MC syndrome' (Malpuech-Michels-Mingarelli-Carnevale syndrome). ... In a review of the literature of similar syndromes, Al Kaissi et al. (2007) concluded that their patients were most similar to those described by Carnevale et al. (1989), Guion-Almeida and Rodini (1995), and Mingarelli et al. (1996), and suggested the designation 'Carnevale syndrome.' The authors noted the similarities to both Michels syndrome and Malpuech syndrome, but considered them to be separate entities.







