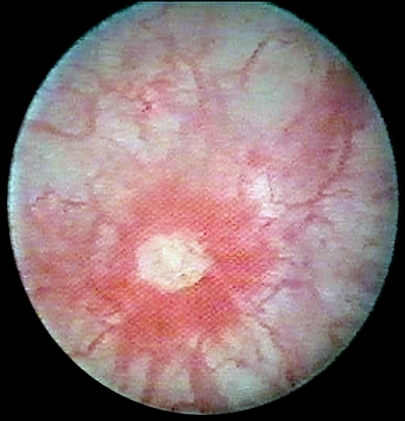
., “Retinal vasculitis occurring with common variable immunodeficiency syndrome,” American Journal of Ophthalmology, vol. 129, no. 2, pp. 269–270, 2000.
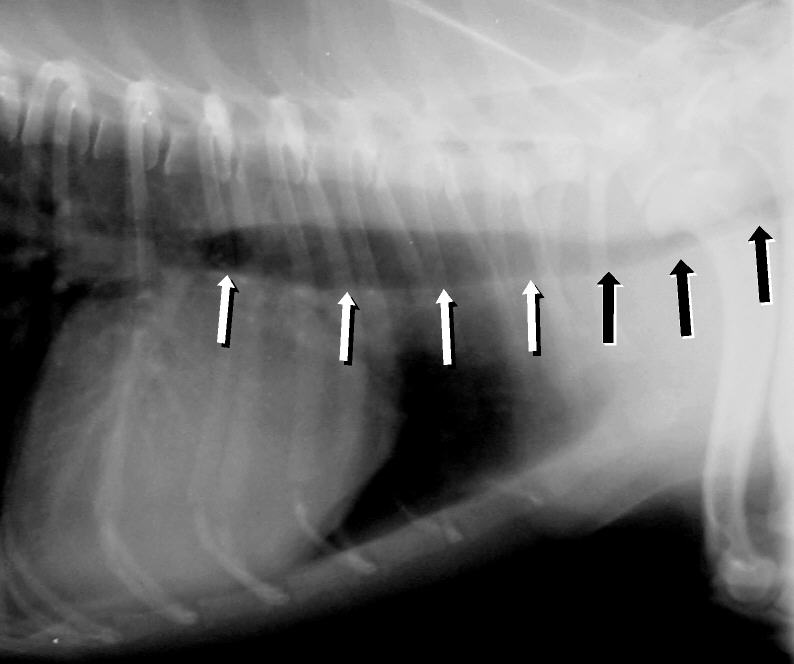
Acquired tracheal collapse can be caused by Cushing's syndrome , heart disease, and chronic respiratory disease and infection. [3] Symptoms include a cough (often called a "goose honk cough" due to its sound), especially when the dog is excited.
Angebault et al. (2015) also studied 2 sisters, born of nonconsanguineous parents, who were 14 and 12 years old, respectively, and had severe bilateral optic neuropathy associated with nystagmus as well as a mild statokinetic cerebellar syndrome and learning disabilities. The older sister was more severely affected, with mild mental retardation and generalized seizures that began at age 3 years.
Two sibs developed acute myeloid leukemia and myelodysplastic syndrome, respectively. Less common features, occurring in only a few patients, included facial dysmorphism, cardiomyopathy or hypertrophy, and hypothyroidism.
A rare organic aciduria characterized by increased urinary excretion of 3-methylglutaconic acid, variably associated with neutropenia (sometimes causing recurrent severe infections and potentially resulting in leukemia) and progressive neurologic manifestations, such as global developmental delay, intellectual disability, hypotonia, movement disorder, and seizures. Microcephaly, cataract, facial dysmorphism, growth retardation, endocrine abnormalities, and cardiomyopathy have also been reported. Brain imaging may show cerebral or cerebellar atrophy, or abnormalities of the basal ganglia.
Three patients also had transient conjugated hyperbilirubinemia during illness, which was attributed to a homozygous S1342Y mutation in the ABCC2 gene (601107), resulting in Dubin-Johnson syndrome (DJS; 237500). Inheritance The transmission pattern of CPSQ3 in the family reported by Kruer et al. (2013) was consistent with autosomal recessive inheritance.
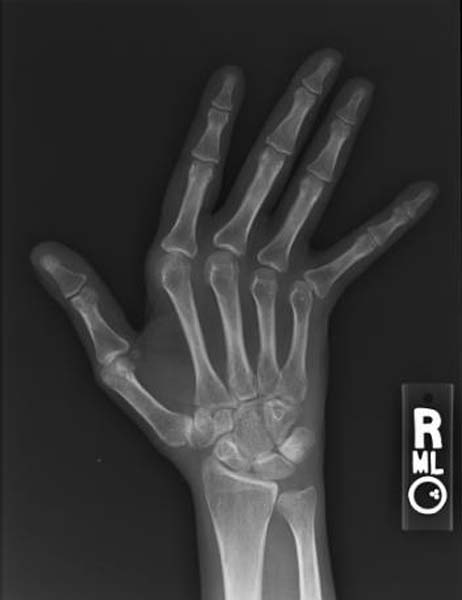
Specialty Rheumatology Jaccoud arthropathy ( JA ), is a chronic non-erosive reversible joint disorder that may occur after repeated bouts of arthritis. [1] [2] It is caused by inflammation of the joint capsule and subsequent fibrotic retraction, causing ulnar deviation of the fingers, through metacarpophalangeal joint (MCP) subluxation, [1] [3] primarily of the ring and little-finger. [3] Joints in the feet, knees and shoulders may also get affected. [1] It is commonly associated with systemic lupus erythematosus (SLE), and occurs in roughly 5% of all cases. [1] [2] When associated with rheumatic fever it is also called chronic post–RF arthropathy . [3] Contents 1 Presentation 1.1 Associated conditions 1.2 Diagnosis 2 Treatment 3 See also 4 References 5 External links Presentation [ edit ] Associated conditions [ edit ] Originally thought to be associated only with rheumatic fever , it has since been shown to occur also in SLE, Sjögren syndrome , scleroderma , dermatomyositis , psoriatic arthritis , vasculitis , ankylosing spondylitis , mixed connective tissue disease , and pyrophosphate deposition disease . [1] It is distinct from bone erosion which is commonly associated with rheumatic arthritis , [1] and also distinct from mild deforming arthropathy which is associated with SLE . [2] There have also been cases of non-rheumatic JA associated with Lyme disease , HIV -infection and a number of other conditions. [1] Diagnosis [ edit ] Plain radiograph hand radiographs typically show marked ulnar subluxation and deviation at the metacarpophalangeal joints.
Differential diagnosis For patients presenting with widespread bullous lesions the differential diagnosis should include bullous congenital ichthyosiform erythroderma, early-onset forms of epidermolysis bullosa, Poikiloderma of Kindler (see these terms) and staphylococcal scalded skin syndrome. Management and treatment Treatment is symptomatic with administration of antihistamines (H1 and H2 in cases with gastrointestinal symptoms), topical steroids and mast cell membrane stabilizers.
Differential diagnosis Differential diagnoses include progressive familial intrahepatic cholestasis, diseases that present with neonatal cholestasis, which includes alpha-1-antitrypsin deficiency of ZZ phenotype, Alagille syndrome, biliary atresia, cystic fibrosis, and metabolic diseases (tyrosinemia type I, galactosemia, hereditary fructose intolerance) (see these terms), diseases that present with fat and fat soluble vitamin malabsorption, including other liver diseases, and intestinal disease, or diseases that present with growth failure.
Congenital bile acid synthesis defect type 1 is a disorder characterized by cholestasis, a condition that impairs the production and release of a digestive fluid called bile from liver cells. Bile is used during digestion to absorb fats and fat-soluble vitamins, such as vitamins A, D, E, and K. People with congenital bile acid synthesis defect type 1 cannot produce (synthesize) bile acids, which are a component of bile that stimulate bile flow and help it absorb fats and fat-soluble vitamins. As a result, an abnormal form of bile is produced. The signs and symptoms of congenital bile acid synthesis defect type 1 often develop during the first weeks of life, but they can begin anytime from infancy into adulthood. Affected infants often have a failure to gain weight and grow at the expected rate (failure to thrive) and yellowing of the skin and eyes (jaundice) due to impaired bile flow and a buildup of partially formed bile.
A number sign (#) is used with this entry because this disorder of bile acid synthesis, referred to here as CBAS1, can be caused by homozygous or compound heterozygous mutation in the gene encoding 3-beta-hydroxy-delta-5-C27-steroid oxidoreductase (HSD3B7; 607764) on chromosome 16p. Description Congenital defects of bile acid synthesis are autosomal recessive disorders characterized by neonatal onset of progressive liver disease with cholestatic jaundice and malabsorption of lipids and lipid-soluble vitamins from the gastrointestinal tract resulting from a primary failure to synthesize bile acids. Affected infants show failure to thrive and secondary coagulopathy. In most forms of the disorder, there is a favorable response to oral bile acid therapy (summary by Cheng et al., 2003). Genetic Heterogeneity of Congenital Defects in Bile Acid Synthesis There are several disorders that result from defects in bile acid synthesis. See CBAS2 (235555), caused by mutation in the delta(4)-3-oxosteroid 5-beta-reductase gene (AKR1D1; 604741) on chromosome 7q33; CBAS3 (613812), caused by mutation in the 7-alpha hydroxylase gene (CYP7B1; 603711) on chromosome 8q12; CBAS4 (214950), caused by mutation in the AMACR gene (604489) on chromosome 5p13; CBAS5 (616278), caused by mutation in the ABCD3 gene (170995) on chromosome 1p21; and CBAS6 (617308), caused by mutation in the ACOX2 gene (601641) on chromosome 3p14.
The affected sibs had normal physical development and body mass index, and exhibited no features of Sensenbrenner syndrome (see CED3, 614099) such as polydactyly, short ribs, or micromelia.
It is a heterogeneous disorder belonging to the group of myelodysplastic/myeloproliferative (MDS/MPN) syndromes. In aCML many clinical features ( splenomegaly , myeloid predominance in the bone marrow with some dysplastic features but without a differentiation block) and laboratory abnormalities (myeloid proliferation, low leukocyte alkaline phosphatase values) suggest the diagnosis of chronic myelogenous leukemia (CML).
A rare myelodysplastic/myeloproliferative neoplasm characterized by peripheral blood leukocytosis due to increased numbers of morphologically dysplastic neutrophils and their precursors, hypercellular bone marrow with granulocytic proliferation and dysplasia (with or without dysplasia in the erythroid and megakaryocytic lineages), and prominent dysgranulopoiesis, but no or minimal absolute basophilia or monocytosis. Blasts account for less than 20% of leukocytes in the blood and bone marrow. BCR-ABL1 fusion is absent, as well as PDGFRA, PDGFRB or FGFR1 rearrangement, or PCM1-JAK2. Patients may present with signs and symptoms related to splenomegaly, anemia, or thrombocytopenia. Prognosis is generally poor.
In an inbred Amish group, Cross et al. (1968) observed 2 sisters with cretinism and the Kocher-Debre-Semelaigne syndrome (myotonia and muscular pseudohypertrophy). ... Because of facial appearance, Aarskog syndrome (305400) had been suggested. The importance of recognizing the true nature of this patient's disorder as congenital hypothyroidism is obvious.
Thyroid hypoplasia is a form of thyroid dysgenesis (see this term) characterized by incomplete development of the thyroid gland that results in primary congenital hypothyroidism (see this term), a permanent thyroid deficiency that is present from birth. Epidemiology Prevalence is estimated at around 1/28,000. Thyroid hypoplasia and athyreosis (see this term) combined account for one-third of cases of thyroid dysgenesis. Clinical description Clinical manifestations of thyroid hypoplasia are often subtle or not present at birth, probably as a result of trans-placental passage of some maternal thyroid hormone or due to the fact that many infants have some thyroid production of their own. More specific symptoms and signs do not develop until several months of age. Common clinical features and signs include decreased activity and increased sleep, feeding difficulty and constipation, prolonged jaundice, myxedematous facies, large fontanels (especially posterior), macroglossia, a distended abdomen with umbilical hernia, and hypotonia.
A number sign (#) is used with this entry because congenital nongoitrous hypothyroidism-5 (CHNG5) is caused by heterozygous mutation in the NKX2-5 gene (600584) on chromosome 5q35. For a general phenotypic description and a discussion of genetic heterogeneity of congenital nongoitrous hypothyroidism, see 275200. Molecular Genetics Dentice et al. (2006) screened for mutations in the coding region of the NKX2-5 gene (600584) in 241 patients with congenital nongoitrous hypothyroidism, including 53 with athyreosis, 99 with thyroid ectopy, and 15 with hypoplasia, and identified 3 different heterozygous missense mutations in 4 of the patients: 2 of the mutations were novel (600584.0015-600584.0016) and the other had previously been identified in patients with congenital heart disease (600584.0004). Functional characterization of the 3 mutations demonstrated reduced DNA binding and/or transactivation properties, with a dominant-negative effect on wildtype NKX2-5. INHERITANCE - Autosomal dominant GROWTH Other - Growth retardation, severe (if untreated) NEUROLOGIC Central Nervous System - Mental retardation, severe (if untreated) ENDOCRINE FEATURES - Hypothyroidism, nongoitrous - Hypoplastic thyroid gland - Ectopic thyroid gland LABORATORY ABNORMALITIES - Increased TSH - Decreased free T(3)/free T(4) MOLECULAR BASIS - Caused by mutation in the E homolog of the Drosophila NK2 transcription factor (NKX2E, 600584.0004 ) ▲ Close
PMID 10607817 . v t e Metal deficiency and toxicity disorders Iron excess: Iron overload Hemochromatosis Hemochromatosis/HFE1 Juvenile/HFE2 HFE3 African iron overload/HFE4 Aceruloplasminemia Atransferrinemia Hemosiderosis deficiency: Iron deficiency Copper excess: Copper toxicity Wilson's disease deficiency: Copper deficiency Menkes disease / Occipital horn syndrome Zinc excess: Zinc toxicity deficiency: Acrodermatitis enteropathica Other Inborn errors of metabolism
Retrieved 2007-02-27 . v t e Skin cancer of nevi and melanomas Melanoma Mucosal melanoma Superficial spreading melanoma Nodular melanoma lentigo Lentigo maligna / Lentigo maligna melanoma Acral lentiginous melanoma Amelanotic melanoma Desmoplastic melanoma Melanoma with features of a Spitz nevus Melanoma with small nevus-like cells Polypoid melanoma Nevoid melanoma Melanocytic tumors of uncertain malignant potential Nevus / melanocytic nevus Nevus of Ito / Nevus of Ota Spitz nevus Pigmented spindle cell nevus Halo nevus Pseudomelanoma Blue nevus of Jadassohn–Tièche Cellular Epithelioid Deep penetrating Amelanotic Malignant Congenital melanocytic nevus ( Giant Medium-sized Small-sized ) Balloon cell nevus Dysplastic nevus / Dysplastic nevus syndrome Acral nevus Becker's nevus Benign melanocytic nevus Nevus spilus This article about a neoplasm is a stub .
Flexion teardrop fractures usually involve instability in all elements of the spine at the injured level, commonly occur at the C4-C7 vertebra, and have a high association with spinal cord injury (in particular anterior cord syndrome ). In comparison, the extension-type fracture occurs more commonly at C2 or C3, causes less if any disruption to the middle and posterior elements, and does not usually result in spinal cord injury (however it may co-occur with more dangerous spine injuries). [4] [5] References [ edit ] ^ eMedicine > Fracture, Cervical Spine By Moira Davenport.
While feline upper respiratory disease can be caused by several different pathogens, there are a few symptoms that they have in common. [1] Avian flu can also infect cats, but "cat flu" is generally a misnomer , since it usually does not refer to an infection by an influenza virus. Instead, it is a syndrome , a term referring to patients displaying a number of symptoms that can be caused by one or more of these infectious agents ( pathogens ): Feline herpes virus causing feline viral rhinotracheitis (cat common cold ).