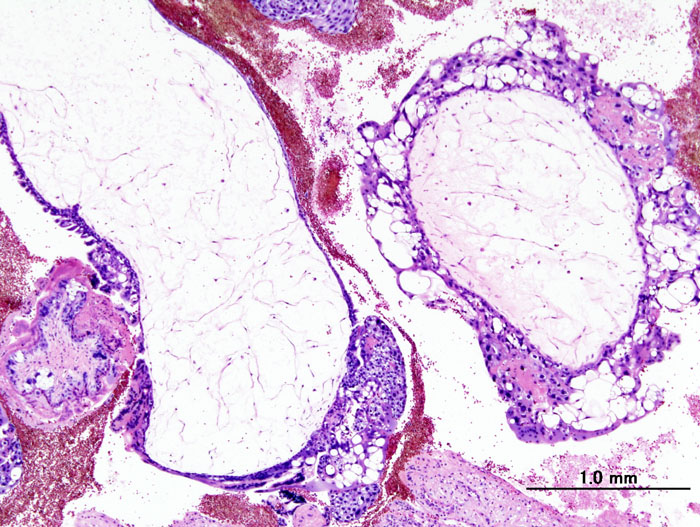
In studies of the Dutch form of hereditary cerebral hemorrhage with amyloidosis, van Duinen et al. (1987) demonstrated that the vascular amyloid deposits were related to the beta-protein associated with Alzheimer disease and Down syndrome (190685). The findings indicated that the 'Dutch type' is genetically distinct from the 'Icelandic type' of cerebroarterial amyloidosis (105150), which is due to a defect in cystatin C (CST3; 604312). ... Of 19 next of kin who survived beyond 60 years of age, 6 had brain disorders; 4 of the 6 presented at least 3 components of the syndrome. The proband's mother had died at age 83 with profound dementia; one sister, who was diagnosed with dementia with occipital calcifications and leukoencephalopathy at age 67, died 2 years later from intracranial hemorrhage; a brother had an occipital hemorrhage at age 58, at which time occipital calcifications and leukoencephalopathy were discovered; and another brother died after a minor stroke at age 70 with dementia, occipital calcifications, and external carotid artery dysplasia. ... Two of the patients had clinical evidence of a dementing syndrome. Ultrastructural studies confirmed the amyloid nature of the congophilic material in the 2 biopsied cases.
A form of hereditary cerebral hemorrhage with amyloidosis characterized by age of onset between 50-66 years of age, memory impairment, myoclonic jerks, expressive dysphagia, short-stepped gait, personality changes, and lobar intracerebral hemorrhages. This subtype is due to a mutation in the APP gene (21q21.2), encoding the beta-amyloid precursor protein. This mutation causes an increased accumulation of amyloid-beta protein in the walls of the arteries and capillaries of the meninges, cerebellar cortex and cerebral cortex, leading to the weakening and eventual rupture of these vessels.
Hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D) is a form of HCHWA (see this term), a group of familial central nervous system disorders, characterized by severe cerebral amyloid angiopathy (CAA), hemorrhagic and non-hemorrhagic strokes and dementia. Epidemiology The prevalence is unknown. It has been seen in three Dutch families, that are most likely related (before 17th century), to date. Approximately 200-300 patients are known to be affected. Clinical description HCHWA-D presents with hemorrhagic stroke between the ages of 39-76 years (average age 50 years). In one third of cases, the initial stroke is fatal, recurrent strokes are seen in those who survive and patients often suffer from serious disability. Cognitive decline can be a presenting symptom in some cases but often occurs after the initial or recurrent strokes.
Hereditary cerebral amyloid angiopathy is a condition that can cause a progressive loss of intellectual function (dementia), stroke, and other neurological problems starting in mid-adulthood. Due to neurological decline, this condition is typically fatal in one's sixties, although there is variation depending on the severity of the signs and symptoms. Most affected individuals die within a decade after signs and symptoms first appear, although some people with the disease have survived longer. There are many different types of hereditary cerebral amyloid angiopathy. The different types are distinguished by their genetic cause and the signs and symptoms that occur.
A form of hereditary cerebral hemorrhage with amyloidosis characterized by an age of onset between 50-70 years of age, recurrent lobar intracerebral hemorrhages and cognitive decline. This subtype is due to a mutation in the APP gene (21q21.2), encoding the beta-amyloid precursor protein. This mutation causes an increased accumulation of amyloid-beta protein in the walls of the arteries and capillaries of the meninges, cerebellar cortex and cerebral cortex, leading to the weakening and eventual rupture of these vessels.
A form of hereditary cerebral hemorrhage with amyloidosis characterized by an age of onset of 45 years of age, progressive Alzheimer's disease-like dementia, and lobar intracerebral hemorrhage in some patients. This subtype is due to a mutation in the APP gene (21q21.2), encoding the beta-amyloid precursor protein. This mutation causes an increased accumulation of amyloid-beta protein in the walls of the arteries and capillaries of the meninges, cerebellar cortex and cerebral cortex, leading to the weakening and eventual rupture of these vessels.
A form of hereditary cerebral hemorrhage with amyloidosis characterized by an age of onset of 54-61 years, progressive Alzheimer's disease-like dementia, and absence of intracerebral hemorrhages. This subtype is due to a mutation in the APP gene (21q21.2), encoding the beta-amyloid precursor protein. This mutation causes an increased accumulation of amyloid-beta protein in the walls of the arteries and capillaries of the meninges, cerebellar cortex and cerebral cortex, leading to the weakening and eventual rupture of these vessels.
A form of hereditary cerebral hemorrhage with amyloidosis characterized by an age of onset of 50 years of age, dementia and lobar intracerebral hemorrhage. This subtype is due to a mutation in the APP gene (21q21.2), encoding the beta-amyloid precursor protein. This mutation causes an increased accumulation of amyloid-beta protein in the walls of the arteries and capillaries of the meninges, cerebellar cortex and cerebral cortex, leading to the weakening and eventual rupture of these vessels.
Her younger sister presented with loss of pain and temperature sensation of both hands, right Horner syndrome and impaired corneal response, and extensor plantar responses. ... One had Chiari type I malformation and syringomyelia, and the other had syringomyelia and Klippel-Feil syndrome (see 118100). The authors hypothesized that genetic factors played a role in the etiology of abnormalities of these hindbrain structures. ... Clinical features included headaches, pseudotumor-like episodes, a Meniere-like syndrome, lower cranial nerve signs, and spinal cord disturbances.
A central nervous system malformation characterized by caudal displacement of the cerebellar tonsils exceeding 5mm below the foramen magnum with or without syringomyelia. Symptoms vary in onset and severity and include suboccipital headache, neck pain, vertigo, tinnitus, ocular symptoms (diplopia, blurred vision, photofobia, nystagmus), lower cranial nerve signs, cerebellar ataxia, and spasticity. Some affected individuals can be asymptomatic.
Find sources: "Nomophobia" – news · newspapers · books · scholar · JSTOR ( December 2016 ) Smartphone use has become ubiquitous throughout many societies Nomophobia (short for 'no mobile phobia') is a humorous word for the fear of, or anxiety caused by, not having a working mobile phone. [1] [2] It has been considered a symptom or syndrome of problematic digital media use in mental health , the definitions of which are not standardized. [3] [4] Contents 1 Overview 2 Research evidence 2.1 Other experiments 3 Symptoms and signs 3.1 Symptoms 3.2 Emotional symptoms 4 Treatments 5 See also 6 References Overview [ edit ] Although nomophobia does not appear in the current Diagnostic and Statistical Manual of Mental Disorders , Fifth Edition (DSM-5), it has been proposed as a "specific phobia," based on definitions given in the DSM-IV. [5] [ dubious – discuss ] According to Bianchi and Philips (2005) psychological factors are involved in the overuse of a mobile phone. [6] These could include low self-esteem (when individuals looking for reassurance use the mobile phone in inappropriate ways) and extroverted personality (when naturally social individuals use the mobile phone to excess). ... About 44% of group 1 reported that they felt "secure" when they had their mobile phones versus 46% of group 2 reported they would not feel the same without their mobile phone. [18] The results demonstrated that 68% of all participants reported mobile phone dependency, but overall the participants with panic disorder and agoraphobia reported significantly more emotional symptoms and dependency on mobile phones when compared to the control group when access to the mobile phone was prohibited. [18] Symptoms and signs [ edit ] Nomophobia occurs in situations when an individual experiences anxiety due to the fear of not having access to a mobile phone. The "over-connection syndrome" occurs when mobile phone use reduces the amount of face-to-face interactions thereby interfering significantly with an individual’s social and family interactions. ... Even though nomophobia is a fairly new concept, there are validated psychometric scales available to help in the diagnostic, an example of one of these scales is the "Questionnaire of Dependence of Mobile Phone/Test of Mobile Phone Dependence (QDMP/TMPD)". [20] See also [ edit ] Mobile phone overuse Screen time Digital media use and mental health Autophobia , fear of being alone Internet addiction disorder Problematic smartphone use Technostress Telephone phobia De Quervain syndrome References [ edit ] ^ a b Charlie D'Agata Nomophobia: Fear of being without your cell phone .
The grade of microtia usually correlates to the degree of development of the middle ear. [9] [15] Microtia is usually isolated, but may occur in conjunction with hemifacial microsomia , Goldenhar Syndrome or Treacher-Collins Syndrome . [26] It is also occasionally associated with kidney abnormalities (rarely life-threatening), and jaw problems, and more rarely, heart defects and vertebral deformities. [18] Notable cases [ edit ] Paul Stanley , vocalist and rhythm guitarist of Kiss , was born with grade III microtia of his right ear. ... PMID 16750771 . ^ Huston Katsanis S, Cutting GR (July 2004). "Treacher Collins Syndrome". GeneReviews . PMID 20301704 .
A congenital malformation of the external ear, seen more frequently in males, that occurs sporadically or is inherited, that is characterized by unilateral (79-93% of cases, 60% of which involve the right ear) or bilateral small and abnormally shaped auricles and that is often associated with atresia or stenosis of the ear canal, attention deficit disorders and delayed language development. The variation in auricle size ranges from grade I, where the auricle is simply smaller than normal, to grade IV, also known as anotia, where there is a complete absence of the external ear and of the auditory canal.
Clinical Features MacCollum (1938) reported a large series but gave no genetic information. Leung et al. (2007) reported a 4-generation Chinese family with isolated bilateral lop ear anomaly. The 6-month-old male proband had superior helical over-folding, absence of the superior (posterior) crus of the antihelix, decreased auricular vertical length, and reduction of the scapha bilaterally. His mother, maternal aunt, maternal grandmother, and maternal great-grandmother had similar abnormalities of both ears. None of the individuals reported hearing problems. Leung et al. (2007) concluded that the pedigree suggested autosomal dominant inheritance.
Overweight The overweight range according to the body mass index (BMI) is the area on the chart where BMI > 25 Specialty Endocrinology Part of a series on Human body weight General concepts Obesity ( Epidemiology ) Overweight Underweight Body shape Weight gain Weight loss Gestational weight gain Diet (nutrition) Weight management Overnutrition Childhood obesity ( Epidemiology ) Medical concepts Adipose tissue Classification of obesity Genetics of obesity Metabolic syndrome ( Epidemiology of metabolic syndrome ) Metabolically healthy obesity Obesity paradox Measurements Body adiposity index Body mass index Body fat percentage Body Shape Index Corpulence index Lean body mass Relative Fat Mass Waist–hip ratio Waist-to-height ratio Related conditions Diabetes ( Type 1 ) Eating disorder ( Anorexia • Bulimia • Binge eating disorder ) Food addiction Hyperthyroidism Malnutrition RED-S Starvation ( Starvation response ) PCOS Obesity-associated morbidity Arteriosclerosis Atherosclerosis Fatty liver disease GERD Heart disease Hypertension Obesity and cancer Osteoarthritis Prediabetes Sleep apnea Type 2 diabetes Management of obesity Anti-obesity medication Bariatrics Bariatric surgery Dieting ( List of diets ) Caloric deficit Exercise ( outline ) Liposuction Obesity medicine Weight loss camp Weight loss coaching Yo-yo effect Social aspects Comfort food Fast food ( Criticism ) Fat acceptance movement Fat fetishism Health at Every Size Hunger Obesity and the environment Sedentary lifestyle Social determinants of obesity Social stigma of obesity Weight cutting Weight class v t e Being overweight or fat is having more body fat than is optimally healthy. ... Temporal Trends 1985–2004 at the Wayback Machine (archived December 8, 2006) Ranking of Most Overweight Countries in the World 2005 at the Wayback Machine (archived June 29, 2007) World Health Organization fact sheet on obesity and overweight v t e Obesity Overweight Childhood obesity Abdominal obesity Weight gain Obesity hypoventilation syndrome Bariatric surgery Obesity and walking Overnutrition
Prontera et al. (2015) considered the proband to have a form of SHORT syndrome (269880), with the additional features of elevated IGF1, central nervous system defects, developmental delay, and a pronounced progeroid appearance, which they designated 'SHORT syndrome type 2.' The authors also noted that distinguishing this phenotype from neonatal progeroid syndrome (264090) could be challenging, especially at birth.
Growth delay due to IGF-I resistance is characterised by variable intrauterine and postnatal growth retardation and elevated serum IGF-I levels. Addition features include variable degrees of intellectual deficit, microcephaly and dysmorphism (broad nasal bridge and tip, smooth philtrum, thin upper and everted lower lips, short fingers, clinodactyly, wide-set nipples and pectus excavatum). Epidemiology Prevalence is unknown. Etiology IGF-I resistance may be caused by a variety of genetic defects: ring chromosome 15, distal heterozygous 15q deletions encompassing the IGF1R gene (15q26.3), or IGF1R gene mutations. Intellectual deficit is pronounced in patients with ring chromosome 15 but varies depending on the size of the deletion and on the functions of other deleted genes in patients with 15q deletions. Partial IGF-I insensitivity due to IGF1R haploinsufficiency has been reported in one patient with a small deletion encompassing one allele of the IGF1R gene and was characterised by small size for gestational age, persistent growth failure that improved considerably with GH therapy, and the absence of intellectual deficit.
Gestational trophoblastic tumors (GTT) are malignant forms of gestational trophoblastic disease. The tumor always follows pregnancy, most often molar pregnancy (hydatidiform mole; see this term). Four histological subtypes have been described: invasive mole, gestational choriocarcinoma, placental site trophoblastic tumor and epithelioid trophoblastic tumor (see these terms). Epidemiology Exact annual incidence is not known but it is estimated to be about 1/1,000,000 women. Clinical description GTTs occur following hydatidiform moles (see this term) (15% of complete moles and about 3% of partial moles), following spontaneous miscarriage (1/150) or childbirth (1/40,000).
A rare, neurodegenerative disease characterized by progressive cataracts, hearing loss, cerebellar ataxia, paranoid psychosis and dementia. Neuropathological features are diffuse atrophy of all parts of the brain, chronic diffuse encephalopathy and the presence of extremely thin and almost completely demyelinated cranial nerves.
A rare, neurodegenerative disease characterized by progressive dementia and ataxia, widespread cerebral amyloid angiopathy and parenchymal amyloid deposition. Two subtypes have been identified, ABri amyloidosis and ADan amyloidosis.
None of the patients had a history of any other type of cancer-prone syndrome or radiation exposure. There was no history of autoimmune disease in the family.
Addison disease can also be one of several features of other genetic conditions, including X-linked adrenoleukodystrophy and autoimmune polyglandular syndrome, type 1, which are caused by mutations in other genes.
Lentigo maligna melanoma (LMM) is a type of skin cancer that usually develops in older, fair-skinned adults. The average age of diagnosis is 65. LMM is thought to be caused by a history of sun exposure to the affected area. Treatment includes surgery to remove as much of the LMM as possible.
Twin studies estimate the heritability of syndromic bulimia to be 54 to 83% (Kendler et al., 1991; Bulik et al., 1998; Wade et al., 1999; Kortegaard et al., 2001).
None of the affected members of this family had any features of growth hormone insensitivity syndrome (262500). Increased Responsiveness to Growth Hormone The GHR gene contains a polymorphism consisting of a genomic deletion of exon 3 (d3GHR) (600946.0031), which mimics alternative splicing.
Short stature due to partial GHR deficiency is a rare, genetic, endocrine disease characterized by idiopathic short stature due to diminished GHR function (decreased ligand binding or reduced availability of receptor), thus resulting in partial insensitivity to growth hormone.
A number sign (#) is used with this entry because of evidence that isolated partial growth hormone (GH; 139350) deficiency (GHDP) is caused by heterozygous, compound heterozygous, or homozygous mutation in the growth hormone secretagogue receptor gene (GHSR; 601898) on chromosome 3q26. Molecular Genetics Pantel et al. (2006) screened for mutations in the GHSR gene in 41 probands with idiopathic short stature and 51 probands with isolated growth hormone deficiency and identified the same mutation (A204E; 601898.0001) in 2 probands of unrelated Moroccan families. No mutations in the GH1 (139250) or GHRHR (139191) genes were identified in the probands. In family 1, which was consanguineous, the proband was homozygous for the mutation; 2 affected sibs and the affected parents were all heterozygous for the mutation, as was an unaffected sib whose height was in the low range of normal (-1.1 SD below the mean). In family 2, the proband and her affected father were both heterozygous for the mutation, as were 2 unaffected sibs, 1 of normal height and the other in the low range of normal (-1.2 SD below the mean), consistent with incomplete penetrance and variable expressivity.
Short stature due to GHSR deficiency is a rare, genetic, endocrine growth disease, resulting from growth hormone secretagogue receptor (GHSR) deficiency, characterized by postnatal growth delay that results in short stature (less than -2 SD). The pituitary gland is typically without morphological changes, although anterior pituitary gland hypoplasia has been reported.
There are many examples of hemizygous selection of differentiating blood cells in the human: in platelets and T cells of women heterozygous for the Wiskott-Aldrich syndrome (301000), in B cells of women heterozygous for agammaglobulinemia (300755), and so on.
Bond et al. (2004) studied 88 individuals who were members of Li-Fraumeni syndrome (LFS1; 151623) families and had germline mutations in 1 allele of p53.
ISBN 978-1-4381-2098-0 . v t e Superstition Main topics Amulet Evil eye Luck Omen Talismans Myth and ritual Lists List of superstitions List of lucky symbols List of bad luck signs Sailors' superstitions Theatrical superstitions Africa Buda Gris-gris Sampy Sleeping child Americas Ascalapha odorata Carranca Cooties Curupira Djucu Fortune cookie Groundhog Day I'noGo tied Oscar love curse Susto White lighter myth Witch window Asia Superstition in India Superstition in Pakistan Japanese superstitions Bhoot (ghost) Chhaupadi Churel Ghosts in Bengali culture Jackal's horn Kuai Kuai culture Muhurta Navaratna Nazar battu Pichal Peri Puppy pregnancy syndrome Akabeko Kanai Anzen Maneki-neko Okiagari-koboshi Omamori Fan death Agimat Arbularyo Barang Kulam Lihi Pagtatawas Pasma Usog Kuman Thong Palad khik Takrut Nang Kwak White elephant Curse of 39 Jin Chan Numbers in Chinese culture Superstitions of Malaysian Chinese Europe August curse Barbary macaques in Gibraltar Bayern-luck Blarney Stone Cimaruta Cornicello The Goodman's Croft Himmelsbrief Icelandic magical staves In bocca al lupo Kitchen witch Klabautermann Mooncalf Nazar Need-fire Painted pebbles Powder of sympathy Rabbit rabbit rabbit Ravens of the Tower of London Russian traditions and superstitions Spilling water for luck The Scottish Play Troll cross Tycho Brahe days Witch post Wolfssegen General 11:11 4 ( Four-leaf clover , Tetraphobia ) 7 ( Seventh son of a seventh son ) 8 9 13 ( Friday the 13th , The Thirteen Club , Thirteenth floor , Triskaidekaphobia ) 108 111 666 ( Number of the Beast ) Ace of spades Auspicious wedding dates Baseball superstition Beginner's luck Black cat Bread and butter Break a leg Chain letter Cramp-ring Curse Davy Jones' Locker Dead man's hand End-of-the-day betting effect Fear of frogs Fear of ghosts First-foot Flying Dutchman Four Eleven Forty Four Gambler's conceit Good luck charm Human sacrifice Jinx Knocking on wood Law of contagion Literomancy Lock of hair Maternal impression Miasma theory Nelson Numismatic charm Penny Rabbit's foot Rainmaking Ship sponsor Shoes on a table Sign of the horns Something old Spilling salt Statue rubbing Three on a match Threshold Toi toi toi 27 Club Wishing well Witch ball Witching hour Related Apotropaic magic Astrology and science Coincidence Debunker Divination Folk religion Fortune-telling Magic and religion Magical thinking Numerology Perceptions of religious imagery in natural phenomena Post hoc ergo propter hoc Traditional medicine Urban legend Jew Muslim This article about a mental disorder is a stub .
MPAs have been studied in autism , Down syndrome , and in schizophrenia . A 2008 meta-analysis found that MPAs are significantly increased in the autistic population. [1] A 1998 study found that 60% of its schizophrenic sample and 38% of their siblings had 6 or more MPAs (especially in the craniofacial area), while only 5% of the control group showed that many. [2] The most often cited MPA, high arched palate, is described in articles as a microform of a cleft palate . [3] Cleft palates are partly attributable to hypoxia. [4] The vaulted palate caused by nasal obstruction and consequent mouth breathing, without the lateralising effect of the tongue, can produce hypoxia at night.