-
Pierson Syndrome
Omim
Description Pierson syndrome is an autosomal recessive disorder comprising congenital nephrotic syndrome with diffuse mesangial sclerosis and distinct ocular abnormalities, including microcoria and hypoplasia of the ciliary and pupillary muscles, as well as other anomalies. ... Mutations in the LAMB2 gene also cause nephrotic syndrome type 5 with or without mild ocular anomalies (NPHS5; 614199). Clinical Features Pierson et al. (1963) reported 2 sisters with congenital nephrotic syndrome and peculiar eye abnormalities. ... Zenker et al. (2004) reported 2 unrelated families, 1 Turkish and 1 Arabic, with congenital nephrotic syndrome and distinct ocular anomalies. ... Laminin beta-2 deficient mice may provide a model for human congenital or idiopathic nephrotic syndromes and can be excluded as the cause of Finnish congenital nephrotic syndrome (256300) because that disorder maps to chromosome 19.
-
Central Cord Syndrome
Wikipedia
Human spinal cord disorder Central cord syndrome Central cord syndrome is the top diagram Specialty Neurology Neurosurgery Central cord syndrome (CCS) is the most common form of cervical spinal cord injury . ... Further indications for surgery include a neurological decline in spinal cord function in stable patients as well as those who require cervical spinal decompression . [11] See also [ edit ] Spinal cord injury Anterior cord syndrome Posterior cord syndrome Brown-Séquard syndrome References [ edit ] ^ Quencer RM, Bunge RP, Egnor M, Green BA, Puckett W, Naidich TP, Post MJ, Norenberg M (1992). "Acute traumatic central cord syndrome: MRI-pathological correlations". ... Harrop (2010). "Traumatic Central Cord Syndrome: Etiology, Management, and Outcomes" . ... External links [ edit ] NINDS Central Cord Syndrome Information Page Classification D ICD - 10 : S14.1 , S24.1 , S34.1 , T09.3 ICD - 9-CM : C1-4 952.03 , C5-7 952.08 , T1-6 952.13 , T7-12 952.18 , Lumbar 952.2 , Sacral 952.3 MeSH : D020210 DiseasesDB : 33409 SNOMED CT : 282787000 External resources eMedicine : pmr/22 v t e Focal lesions of the spinal cord General Myelopathy Myelitis Spinal cord compression By location Brown-Séquard syndrome Posterior cord syndrome Anterior cord syndrome Central cord syndrome Cauda equina syndrome Other Polio Demyelinating disease Transverse myelitis Tropical spastic paraparesis Epidural abscess Syringomyelia Syringobulbia Morvan's syndrome Sensory ataxia Tabes dorsalis Abadie's sign Subacute combined degeneration of spinal cord Vascular myelopathy Anterior spinal artery syndrome Foix–Alajouanine syndrome

-
Leigh Syndrome With Nephrotic Syndrome
Orphanet
A rare, genetic neurometabolic disease characterized by encephalomyopathy (including developmental delay, nystagmus, progressive ataxia, dystonia, amyotrophy, visual loss, sensorineural deafness, seizures) and bilateral, symmetrical lesions in the basal ganglia or brainstem on imaging, associated with nephrotic syndrome.

-
Pacemaker Syndrome
Wikipedia
Pacemaker syndrome Ventricular pacemaker with 1:1 retrograde ventriculoatrial (V-A) conduction to the atria (arrows). ... Complications [ edit ] Studies have shown that patients with Pacemaker syndrome and/or with sick sinus syndrome are at higher risk of developing fatal complications that calls for the patients to be carefully monitored in the ICU . ... However several risk factors are associated with pacemaker syndrome. [5] [10] Risk factors [ edit ] In the preimplantation period, two variables are predicted to predispose to the syndrome. ... "The pacemaker syndrome -- a matter of definition". Am. ... PMID 1334465 . ^ a b c d "Pacemaker Syndrome: Treatment & Medication - eMedicine Cardiology" . 2018-04-22.
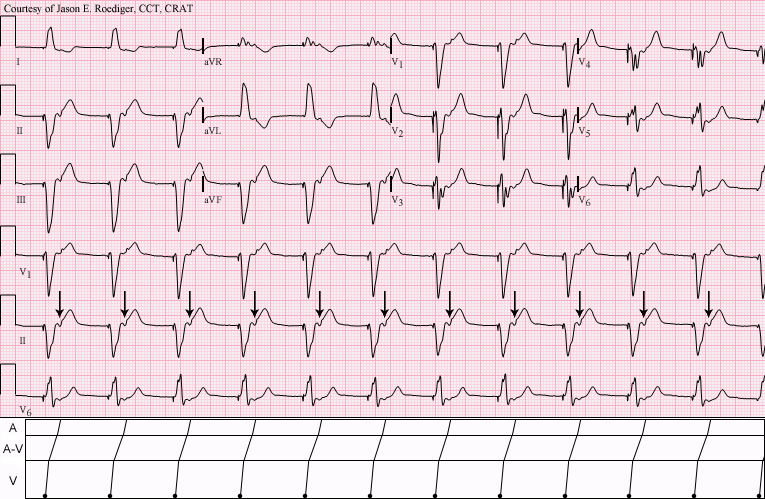
-
Toxic Anterior Segment Syndrome
Wikipedia
Find sources: "Toxic anterior segment syndrome" – news · newspapers · books · scholar · JSTOR ( February 2014 ) ( Learn how and when to remove this template message ) Toxic anterior segment syndrome Specialty Ophthalmology Toxic anterior segment syndrome is an acute, sterile anterior segment inflammation following generally uneventful cataract and anterior segment surgery. [1] One of the main factors in differentiating toxic anterior segment syndrome from an infectious endophthalmitis is the rapid onset. Most patients with toxic anterior segment syndrome will develop symptoms within 12 to 24 hours of the surgery. ... Patients with toxic anterior segment syndrome will often respond rapidly to treatment with topical corticosteroids , while infectious endophthalmitis must be treated with antibiotics . ... Most patients reported to date are in the category of a moderate toxic inflammation. Toxic anterior segment syndrome may be related to problems with any irrigating solution or other solution placed in the patient's eye during surgery, including balanced salt solution or anything added to solutions. ... References [ edit ] ^ "Toxic Anterior Segment Syndrome After Cataract Surgery" . Centers for Disease Control and Prevention . 29 June 2007 .
-
Collagen Disease
Wikipedia
National Cancer Institute document: "Dictionary of Cancer Terms" . v t e Diseases of collagen , laminin and other scleroproteins Collagen disease COL1 : Osteogenesis imperfecta Ehlers–Danlos syndrome, types 1, 2, 7 COL2 : Hypochondrogenesis Achondrogenesis type 2 Stickler syndrome Marshall syndrome Spondyloepiphyseal dysplasia congenita Spondyloepimetaphyseal dysplasia, Strudwick type Kniest dysplasia (see also C2/11 ) COL3 : Ehlers–Danlos syndrome, types 3 & 4 Sack–Barabas syndrome COL4 : Alport syndrome COL5 : Ehlers–Danlos syndrome, types 1 & 2 COL6 : Bethlem myopathy Ullrich congenital muscular dystrophy COL7 : Epidermolysis bullosa dystrophica Recessive dystrophic epidermolysis bullosa Bart syndrome Transient bullous dermolysis of the newborn COL8: Fuchs' dystrophy 1 COL9: Multiple epiphyseal dysplasia 2, 3, 6 COL10: Schmid metaphyseal chondrodysplasia COL11: Weissenbacher–Zweymüller syndrome Otospondylomegaepiphyseal dysplasia (see also C2/11 ) COL17: Bullous pemphigoid COL18: Knobloch syndrome Laminin Junctional epidermolysis bullosa Laryngoonychocutaneous syndrome Other Congenital stromal corneal dystrophy Raine syndrome Urbach–Wiethe disease TECTA DFNA8/12, DFNB21 see also fibrous proteins Authority control NDL : 00566160 This article about a disease of musculoskeletal and connective tissue is a stub .TNF, COL2A1, HLA-B, COL1A1, CRP, HLA-C, IL6, HLA-DRB1, ERAP1, COL1A2, VDR, TTN, TRAF1, ACR, TPO, TNP1, TG, TRBV20OR9-2, TRIM21, TNFAIP3, WNT5A, VEGFA, VIL1, SELE, CDR3, PDLIM7, ADIPOQ, ADAMTS2, IRAK3, PTPN22, ICOS, IL22, FAM167A, IL17F, SOX9, MGP, CCL5, FCGR3A, CXCR5, C2, DDR1, TNFRSF8, CMKLR1, CR2, DES, ELANE, EMP1, ETS1, F2RL1, GRK6, SAA1, GRN, HMGB1, IL1A, IL1B, IL6R, IL17A, LRP5, MAS1, APOH, NT5E, PSMB9, NLRP3
-
Gerodermia Osteodysplastica
Wikipedia
"Geroderma osteodysplasticum hereditaria and wrinkly skin syndrome in 22 patients from Oman". Am. ... "Gerodermia osteodysplastica and wrinkly skin syndrome: are they the same?". Am. J. Med. ... "Gerodermia osteodysplastica/wrinkly skin syndrome: report of three patients and brief review of the literature". ... "Gerodermia osteodysplastica in a Bedouin sibship: further delineation of the syndrome". Clin. Dysmorphol . 6 (1): 51–55. doi : 10.1097/00019605-199701000-00009 . ... External links [ edit ] Classification D ICD - 10 : Q82.9 ICD - 9-CM : 710.9 OMIM : 231070 MeSH : C537799 C537799, C537799 DiseasesDB : 32101 External resources Orphanet : 2078 v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation Birthmark

-
Crome Syndrome
Wikipedia
Crome syndrome Other names Cataract-nephropathy-encephalopathy syndrome [1] Specialty Neurology Crome syndrome is a rare disease defined by various symptoms, including epilepsy , intellectual disability , eye and kidney problems. ... "Orphanet: Cataract nephropathy encephalopathy syndrome" . www.orpha.net . Retrieved 29 June 2019 . ^ "Crome syndrome" . ... Retrieved 2009-04-02 . ^ "Crome syndrome" . Johns Hopkins University . Retrieved 2009-04-02 .
-
Orofaciodigital Syndrome 1
Gard
Orofaciodigital syndrome 1 (OFD1), also called orofaciodigital syndrome type 1, is a condition that affects the development of the oral cavity (the mouth and teeth), facial features, and digits (fingers and toes). This condition also causes polycystic kidney disease. Orofaciodigital syndrome 1 is caused by a change ( mutation) in a gene called OFD1 which appears to play an important role in the early development of many parts of the body including the brain, face, limbs, and kidneys. The syndrome is inherited in an X-linked dominant pattern. ... Researchers have identified at least 13 potential forms of orofaciodigital syndromes, which are classified by their patterns of signs and symptoms. OFD1 is the most common form of orofaciodigital syndrome and differs from the other types mainly by its association with polycystic kidney disease .
-
Poland Syndrome
Medlineplus
The bones of the forearm (radius and ulna) are shortened in some people with Poland syndrome, but this shortening may also be difficult to detect unless measured. ... Rarely, chest and hand abnormalities resembling those of Poland syndrome occur on both sides of the body, but researchers disagree as to whether this condition is a variant of Poland syndrome or a different disorder. Frequency Poland syndrome has been estimated to occur in 1 in 20,000 newborns. ... Causes The cause of Poland syndrome is unknown. Researchers have suggested that it may result from a disruption of blood flow during development before birth. ... Rare cases of Poland syndrome are thought to be caused by a genetic change that can be passed down in families, but no related genes have been identified.
-
Cavernous Venous Malformation
Wikipedia
External links [ edit ] Classification D OMIM : 116860 v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteins
-
Kearns-Sayre Syndrome
Medlineplus
Kearns-Sayre syndrome is a condition that affects many parts of the body, especially the eyes. The features of Kearns-Sayre syndrome usually appear before age 20, and the condition is diagnosed by a few characteristic signs and symptoms. ... People with Kearns-Sayre syndrome may also experience muscle weakness in their limbs, deafness, kidney problems, or a deterioration of cognitive functions (dementia). ... Frequency The prevalence of Kearns-Sayre syndrome is approximately 1 to 3 per 100,000 individuals. ... The mtDNA deletions that cause Kearns-Sayre syndrome result in the loss of genes important for mitochondrial protein formation and oxidative phosphorylation.
-
Mcleod Neuroacanthocytosis Syndrome
Medlineplus
McLeod neuroacanthocytosis syndrome is primarily a neurological disorder that occurs almost exclusively in boys and men. ... People with McLeod neuroacanthocytosis syndrome also have abnormal star-shaped red blood cells (acanthocytosis). ... The signs and symptoms of McLeod neuroacanthocytosis syndrome usually begin in mid-adulthood. ... Frequency McLeod neuroacanthocytosis syndrome is rare; approximately 150 cases have been reported worldwide. ... It is not known how the lack of XK protein leads to the movement problems and other features of McLeod neuroacanthocytosis syndrome. Learn more about the gene associated with McLeod neuroacanthocytosis syndrome XK Inheritance Pattern McLeod neuroacanthocytosis syndrome is inherited in an X-linked recessive pattern .
-
Lowe Syndrome
Medlineplus
Lowe syndrome is a condition that primarily affects the eyes, brain, and kidneys. ... Kidney (renal) abnormalities, most commonly a condition known as renal Fanconi syndrome, frequently develop in individuals with Lowe syndrome. ... In individuals with renal Fanconi syndrome, the kidneys are unable to reabsorb important nutrients into the bloodstream. ... Frequency Lowe syndrome is an uncommon condition. It has an estimated prevalence of 1 in 500,000 people. ... Learn more about the gene associated with Lowe syndrome OCRL Inheritance Pattern This condition is inherited in an X-linked pattern.OCRL, INPP5B, CLCN5, ACTB, ARHGAP1, PHETA1, INPP5E, INPP5K, INPP5D, SCRN1, SLC22A8, APPL1, ABCC4, SLC22A6, SIX2, PALLD, NR1H4, NBAS, BFAR, SNX9, SHH, MCOLN1, PTBP2, SESN2, SCRN2, PIFO, PHETA2, ASPM, SMARCA1, PTEN, SCN7A, RAB5A, AFP, ANXA2, CES1, CETP, ABCC2, CTSD, DCX, G6PD, GH1, GRB2, HOXD13, HPRT1, HTC2, INPP5A, MTM1, NAGLU, PAFAH1B1, PIP, PLEK, PTBP1, ADRB2, ARSH
-
Otopalatodigital Syndrome Type 2
Medlineplus
Otopalatodigital syndrome type 2 is a disorder primarily involving abnormalities in skeletal development. It is a member of a group of related conditions called otopalatodigital spectrum disorders, which also includes otopalatodigital syndrome type 1, frontometaphyseal dysplasia, Melnick-Needles syndrome, and terminal osseous dysplasia. ... Males with otopalatodigital syndrome type 2 generally have much more severe signs and symptoms compared to affected females. ... Its specific incidence is unknown. Causes Otopalatodigital syndrome type 2 is caused by mutations in the FLNA gene. ... The FLNA gene mutations that cause otopalatodigital syndrome type 2 all result in changes to the filamin A protein in the region that binds to actin.
-
Blue Rubber Bleb Nevus Syndrome
Wikipedia
Blue rubber bleb nevus syndrome Other names BRBNS, or Blue rubber bleb syndrome, or Blue rubber-bleb nevus or Bean syndrome The cutaneous vascular malformations of blue rubber bleb nevus syndrome. ... Blue rubber bleb nevus syndrome is difficult to diagnose because of how rare the disease is. ... "Blue rubber bleb naevus syndrome: a rare cause of chronic occult blood loss and iron deficiency anaemia" . ... (January 2017). "Blue Rubber Bleb Nevus (BRBN) Syndrome Is Caused by Somatic TEK (TIE2) Mutations" . ... Retrieved 2018-10-08 . ^ "Blue rubber bleb nevus syndrome (Bean syndrome)" . www.dermatologyadvisor.com .

-
Chilaiditi Syndrome
Wikipedia
"Chilaiditi's syndrome: what should every surgeon know?". ... "Severe Recurrent Abdominal Pain: An Anatomical Variant of Chilaiditi's Syndrome" . MedGenMed . 9 (2): 67. PMC 1994890 . ... "Transverse colon volvulus and associated Chilaiditi's syndrome: case report and literature review". ... PMID 8946999 . ^ a b Keles S, Artac H, Reisli I, Alp H, Koc O (June 2006). "Chilaiditi syndrome as a cause of respiratory distress". ... PMID 16489467 . ^ Walsh SD, Cruikshank JG (February 1977). "Chilaiditi syndrome" . Age Ageing . 6 (1): 51–7. doi : 10.1093/ageing/6.1.51 .

-
Congenital Contractural Arachnodactyly
Wikipedia
Congenital contractual arachnodactyly Other names Beals syndrome; Beals–Hecht syndrome; Arachnodactyly, contractural Beals type; multiple with arachnodactyly; Ear anomalies-contractures-dysplasia of bone with kyphoscoliosis; Distal arthrogryposis type 9 Symptoms Tall, slender body; arm span exceeds height; long, slender fingers and toes; kyphoscoliosis; crumpled ear; joint stiffness Usual onset Conception Causes Mutation of FBN2 gene Treatment Physical therapy for joint contractures; bracing and/or surgical correction for kyphoscoliosis Prognosis Life expectancy depends on severity of symptoms but typically it is not shortened Congenital contractural arachnodactyly ( CCA ), also known as Beals-Hecht syndrome , is a rare autosomal dominant congenital connective tissue disorder. [1] As with Marfan syndrome , people with CCA typically have an arm span that is greater than their height and very long fingers and toes . [2] However, Beals and Hecht discovered in 1972 that, unlike Marfan's, CCA is caused by mutations to the fibrillin-2 ( FBN2 ) gene rather than the fibrillin-1 ( FBN1 ) gene. [1] [3] Contents 1 Signs and symptoms 2 Causes 3 Diagnosis 4 Management 5 Prognosis 6 See also 7 References 8 External links Signs and symptoms [ edit ] CCA is characterized by contractures of varying degrees, mainly involving the large joints, which are present in all affected children at birth. [1] The contractures may be mild and tend to improve over time, but permanently bent fingers and toes ( camptodactyly ) are almost always present. [1] [4] In addition to long fingers and toes and a tall, slender body, people with CCA often have ears that appear to be crumpled , joint stiffness and underdeveloped muscles (muscular hypoplasia ), and they may have curved spines (congenital kyphoscoliosis ). [1] [2] If kyphoscoliosis is present, it often becomes progressively worse and may require surgery. [2] [5] In some cases, the blood vessel that distributes blood from the heart to the rest of the body ( aorta ) may be abnormally enlarged ( aortic root dilatation ). [4] Causes [ edit ] Congenital contractural arachnodactyly may be the result of new mutations in the FBN2 gene or it may be inherited from a parent in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. [2] Congenital contractural arachnodactyly is inherited in an autosomal dominant pattern. ... PMID 20301560 . ^ Hecht, F.; Beals, R. K. (April 1972). " " New" syndrome of congenital contractural arachnodactyly originally described by Marfan in 1896". ... "Congenital contractural arachnodactyly (Beals syndrome)" . Orphanet J Rare Dis . 1 : 20. doi : 10.1186/1750-1172-1-20 . ... External links [ edit ] Classification D ICD - 10 : Q87.8 OMIM : 121050 MeSH : C536211 DiseasesDB : 29326 External resources GeneReviews : Congenital Contractural Arachnodactyly GARD : congenital-contractural-arachnodactyly Orphanet : 115 v t e Systemic connective tissue disorders General Systemic lupus erythematosus Drug-induced SLE Libman–Sacks endocarditis Inflammatory myopathy Myositis Dermatopolymyositis Dermatomyositis / Juvenile dermatomyositis Polymyositis * Inclusion body myositis Scleroderma Systemic scleroderma Progressive systemic sclerosis CREST syndrome Overlap syndrome / Mixed connective tissue disease Other hypersensitivity / autoimmune Sjögren syndrome Other Behçet's disease Polymyalgia rheumatica Eosinophilic fasciitis Eosinophilia–myalgia syndrome fibrillin Marfan syndrome Congenital contractural arachnodactyly v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteinsFBN2, TNNI2, ECEL1, FBN1, AKT1, EGFR, PIK3CA, IL6, MIR21, PIK3CG, PIK3CD, PIK3CB, ERBB2, BCL2, CD274, PTGS2, TNF, IDH1, HDAC3, STAT3, TP53, EZH2, CASP3, CCK, BAP1, ABCB1, YAP1, MCL1, ELAVL2, KRAS, IDH2, CTNNB1, CEACAM5, VEGFA, VIM, TGFB1, FGFR2, PTK2B, GDE1, NFE2L2, VDR, SMAD4, TNFSF10, FBXW7, KRT19, IL10, PPFIBP2, CEACAM7, FOXM1, CEACAM3, MTCO2P12, PSG2, S100A9, GATA6, POSTN, NEAT1, PVT1, PSMD10, COX2, ARID1A, MAPK3, PRKAR1A, GABPA, MET, RECK, IFI27, MMP9, HMOX1, IFNG, HSP90AA1, TP73-AS1, LDHA, LCN2, SLC7A5, RIPK1, CXCR4, ABCC3, AFAP1-AS1, SNAI1, MTHFR, CBX5, SOX4, S100A8, ROS1, STK11, CD163, MAP2K7, MAPK1, PPP3CA, PPIA, POMC, PLK1, DICER1, TFF2, MUC1, TGFBR2, SOCS3, TLR4, SMUG1, SNHG1, PDGFRB, TP73, PDGFA, SERPINB2, PROM1, NR1H4, NOTCH1, OPCML, SPP1, MIR34A, NQO1, CCAT1, MIR485, CD44, MIR494, MIR200A, MIR140, MIR192, DPYD, CDK4, MIR186, BAX, MIR122, EPHB2, CASP9, MIR150, CCND1, CDH1, BSG, FGFR1, AKT2, SOX2-OT, NNT-AS1, MIR210, GGTLC5P, POU5F1P4, UCA1, HTATIP2, ZNF423, TBC1D9, XRCC6P5, MIR424, MIR370, SIRT3, AGR2, ZNRD2, KAT5, SEMA4D, TACC3, GPNMB, DCTN6, CCL27, SIRT2, MIR378A, FILIP1L, CILK1, MIR490, ZHX1, MORC2, ANXA10, POTEM, POU5F1P3, ACOT7, WDHD1, PSIP1, METAP2, FLVCR1-DT, KCNQ1OT1, MALT1, KLRK1, RASSF1, ACTBL2, PAK4, MSLN, SYCE1L, LOXL1-AS1, LMCD1-AS1, TMED7-TICAM2, F2RL3, MCM3AP, SPHK1, APLN, MICA, NRP1, CES2, TNFRSF10C, KLRC4-KLRK1, SPRY4-IT1, PSC, OPCML-IT1, EED, PGR-AS1, PANDAR, CUL4B, CCAT2, CDR1-AS, H3P23, MIR877, MIR876, GGTLC4P, DCAF1, TSPAN1, SRA1, POTEKP, MIR612, FGF19, KLHL21, SETDB1, MAML1, MIR622, SETD1A, ABCG1, MTA1, TP53I3, PTGES, FHL5, MIR637, GGTLC3, S1PR2, GGT2, PTTG1, TMSB10, HULC, MIR551B, MIR25, MIR99A, MIR106B, ARHGAP24, LPAL2, FENDRR, LINC01061, MIRLET7C, MIR106A, TET1, PDGFD, ASRGL1, ZNF703, NEIL1, HSDL2, MIR10A, ZSCAN18, HIF3A, SOX17, PDF, AFAP1, SELENOK, KMT2C, HAMP, KLHL1, C3P1, RPAIN, MIR30E, GATA5, FFAR4, RAB7B, TRIM59, PWAR4, DDX53, GPBAR1, PDIK1L, IL34, PRIMA1, LINC00261, OSCP1, DPY30, LRG1, OSBPL8, OSBPL7, DNER, WDR20, IL33, MIR22HG, TICAM2, LINC-PINT, ACCS, NR0B2, DANCR, MIR126, WWTR1, ERO1A, MIR195, MCTS1, MCAT, PDLIM3, MIR200B, PDCD4, SALL3, ATRNL1, MIR200C, DNAJB5, MIR130A, OSBP2, PLA2G15, MIR203A, SLC7A11, MIR221, CA14, MACC1, SUZ12, MIR29A, MIR30D, IL22, TMED7, PI15, PRLH, MIR132, ACKR3, ACSS2, MEG3, QRSL1, CCDC25, NAT10, PBRM1, MIR142, MIR144, AKIRIN2, TUG1, RNF43, CDHR2, MIR15A, TREM1, SRRT, MIR191, SIRT7, ZBTB7A, TNNI3K, AICDA, NAT1, HMGA2, GLS, GGT1, GATA1, FUT1, FKBP4, FGFR4, FGF10, FGF7, FCGR3B, FCGR3A, FBP1, FABP4, F2RL1, F2R, ETV4, ESD, EREG, ERBB4, SLC29A1, ENO1, EMP1, ELK1, EIF4EBP1, EFNA1, ECM1, EBF1, GPC3, GNAS, DNMT1, GNB3, IL13, CXCL8, IL1B, IGFBP7, IGF2, IGF1, ICAM1, DNAJB1, HES1, AGFG1, HOXD9, HNRNPK, ONECUT1, HMGA1, HK2, HIF1A, HHEX, HGF, HGD, HDAC2, GTF2H4, GSK3B, GRN, GPT, GP2, E2F2, CYP19A1, PDX1, BRAF, ATHS, ASS1, ASPH, APRT, BIRC5, XIAP, APEX1, ANXA5, ANXA1, ANG, ALPP, ALOX15, ALOX5, ALDH1A3, ALB, AGXT, APLNR, AGTR2, PARP1, ACTN4, ACTG2, ACTG1, ACTB, ABCA1, SERPINA3, BCL9, CASP8, CYP1A2, CAT, CTSL, CTSB, CSNK2B, CSF1R, CRYBB2, COL11A2, COL6A3, COL1A2, COL1A1, PLK3, ABCC2, CMA1, CLTC, CHI3L1, CDX2, CDKN2A, CDKN1A, CDK7, CDK1, CD47, CD14, CCT, CCND2, CCNB1, RUNX3, IL17A, ITGB4, MFAP5, STAT1, SOX9, SOX2, SOD2, SLPI, SLC22A3, SLC22A1, SLC16A1, SLC15A1, SLC4A1, SLC3A2, SKP2, SFRP1, CXCL12, SCP2, S100A6, S100A2, ROBO2, RARG, RARB, RAC1, PVR, PTPN11, PTPN6, PTEN, PSMD9, NAT2, TAC1, PRKAR2B, ADAM17, NR4A3, PLA2G7, RAB7A, PRDM2, XRCC1, VCL, UNG, UCHL1, TYMS, TXN, TWIST1, TPT1, TPM1, TPD52, TIMP3, TGFB2, TFRC, TFF3, TFF1, TF, TERC, TEK, TCF21, ZEB1, TACR1, MAPK8, PPARG, ITPR3, GADD45B, MUTYH, MUC5AC, MUC2, NUDT1, MST1R, MRC1, KMT2A, MGMT, MFAP1, MEN1, MARCKS, EPCAM, TACSTD2, LUM, LTA, LOXL2, LOX, LGALS3, LGALS1, LCK, LASP1, RPSA, LAMC2, LAMP1, KDR, MYC, NF2, POU5F1, NFKB2, POU2F1, POLD1, SEPTIN4, PLG, PLAU, PLA2G4A, PKM, PKD2, PEG3, PECAM1, PDR, PDK3, PDGFRA, PDCD1, PCNA, PAWR, PAK3, SERPINE1, PAFAH1B1, ORM2, OGG1, NTS, YBX1, NOTCH3, NOS3, H3P42
-
Liebenberg Syndrome
Medlineplus
Liebenberg syndrome is a condition that involves abnormal development of the arms, resulting in characteristic arm malformations that can vary in severity. ... Frequency Liebenberg syndrome is a rare condition. Fewer than 10 affected families have been described in the medical literature. Causes Liebenberg syndrome is caused by genetic changes near the PITX1 gene. ... The genetic changes involved in Liebenberg syndrome delete, insert, or rearrange genetic material near the PITX1 gene. ... Because the PITX1 protein normally directs lower limb structure, bones, muscles, and tendons in the arms and hands develop more like those in the legs and feet, leading to the features of Liebenberg syndrome. Learn more about the gene associated with Liebenberg syndrome PITX1 Inheritance Pattern Liebenberg syndrome is inherited in an autosomal dominant pattern , which means having a genetic change that affects the PITX1 gene on one copy of the chromosome in each cell is sufficient to cause the disorder.
-
3q29 Microduplication Syndrome
Medlineplus
3q29 microduplication syndrome (also known as 3q29 duplication syndrome) is a condition that results from the copying (duplication ) of a small piece of chromosome 3 in each cell. ... Frequency 3q29 microduplication syndrome appears to be very rare. Fewer than 30 affected individuals have been described in the medical literature. Causes Most people with 3q29 microduplication syndrome have an extra copy of about 1.6 million DNA building blocks (base pairs), also written as 1.6 megabases (Mb), at position q29 on chromosome 3. ... (A missing copy of this segment causes another condition called 3q29 microdeletion syndrome.) The chromosome segment most commonly duplicated in people with 3q29 microduplication syndrome contains about 20 genes. ... However, it is unknown which specific genes, when abnormally copied, are related to the varied signs and symptoms of 3q29 microduplication syndrome. It is also unclear why some people with a duplication at 3q29 have no associated health problems.





