-
Townes-Brocks Syndrome
Medlineplus
Townes-Brocks syndrome is a genetic condition that affects several parts of the body. ... Other signs and symptoms of Townes-Brocks syndrome can include kidney abnormalities, mild to profound hearing loss, eye abnormalities, heart defects, foot abnormalities, and genital malformations. ... Mild intellectual disability or learning problems have been reported in about 10 percent of people with Townes-Brocks syndrome. Frequency The prevalence of this condition is unknown. It is difficult to determine how frequently Townes-Brocks syndrome occurs because the varied signs and symptoms of this disorder overlap with those of other genetic syndromes. Causes Mutations in the SALL1 gene cause Townes-Brocks Syndrome. This gene provides instructions for making a protein that is involved in development before birth.
-
Dnmt3a Overgrowth Syndrome
Medlineplus
DNMT3A overgrowth syndrome is a disorder characterized by faster than normal growth before and after birth, subtle differences in facial features, and intellectual disability. Individuals with DNMT3A overgrowth syndrome are often longer than normal at birth and are taller than their peers throughout life. ... Frequency The prevalence of DNMT3A overgrowth syndrome is unknown. More than 20 affected individuals have been described in the medical literature. Causes As its name suggests, mutations in the DNMT3A gene cause DNMT3A overgrowth syndrome. The DNMT3A gene provides instructions for making an enzyme called DNA methyltransferase 3 alpha. ... Some DNMT3A gene mutations that cause DNMT3A overgrowth syndrome lead to a decrease in normal enzyme function.
-
Allopurinol Hypersensitivity Syndrome
Wikipedia
Allopurinol hypersensitivity syndrome Allopurinol Allopurinol hypersensitivity syndrome typically occurs in persons with preexisting kidney failure . [1] : 119 Weeks to months after allopurinol is begun, the patient develops a morbilliform eruption [1] : 119 or, less commonly, develops one of the far more serious and potentially lethal severe cutaneous adverse reactions viz., the DRESS syndrome , Stevens Johnson syndrome , or toxic epidermal necrolysis . [2] See also [ edit ] Severe cutaneous adverse reactions (i.e. ... External links [ edit ] Classification D ICD - 10 : Y54.8 ICD - 9-CM : E944.7 v t e Adverse drug reactions Antibiotics Penicillin drug reaction Sulfonamide hypersensitivity syndrome Urticarial erythema multiforme Adverse effects of fluoroquinolones Red man syndrome Jarisch–Herxheimer reaction Hormones Steroid acne Steroid folliculitis Chemotherapy Chemotherapy-induced acral erythema Chemotherapy-induced hyperpigmentation Scleroderma-like reaction to taxanes Hydroxyurea dermopathy Exudative hyponychial dermatitis Anticoagulants Anticoagulant-induced skin necrosis Warfarin necrosis Vitamin K reaction Texier's disease Immunologics Adverse reaction to biologic agents Leukotriene receptor antagonist-associated Churg–Strauss syndrome Methotrexate-induced papular eruption Adverse reaction to cytokines Other drugs Anticonvulsant hypersensitivity syndrome Allopurinol hypersensitivity syndrome Vaccine adverse event Eczema vaccinatum Bromoderma Halogenoderma Iododerma General Skin and body membranes Acute generalized exanthematous pustulosis Bullous drug reaction Drug-induced acne Drug-induced angioedema Drug-related gingival hyperplasia Drug-induced lichenoid reaction Drug-induced lupus erythematosus Drug-induced nail changes Drug-induced pigmentation Drug-induced urticaria Stevens–Johnson syndrome Injection site reaction Linear IgA bullous dermatosis Toxic epidermal necrolysis HIV disease-related drug reaction Photosensitive drug reaction Other Drug-induced pseudolymphoma Fixed drug reaction Serum sickness-like reaction This cutaneous condition article is a stub .

-
Streff Syndrome
Wikipedia
Unsourced or poorly sourced material may be challenged and removed . Find sources: "Streff syndrome" – news · newspapers · books · scholar · JSTOR ( June 2020 ) Streff syndrome is a vision condition primarily exhibited by children under periods of visual or emotional stress . ... John Streff as Non-malingering syndrome. In 1962, Dr. Streff and Dr. Richard Apell expanded the concept to add early adaptive syndrome as a precursor to Streff syndrome. ... Some research indicates that Streff syndrome may be caused by a dysfunction in the magnocellular pathway of the retinal ganglion cells . [1] These cells are only 10% of the retinal nerve cells and register motion detection . Early Adaptive Syndrome Diagnosis [ edit ] The diagnostic criteria for Streff syndrome are not well established, and the validity of this condition has not been recognized by The American Academy of Ophthalmology, The American Academy of Pediatric Ophthalmology, The American Academy of Optometry or The American academy of Pediatrics. Treatment [ edit ] Most optometrists agree that Streff syndrome is a generalized reduction in visual performance that is not caused by structural damage.
-
Purple Glove Syndrome
Wikipedia
Purple glove syndrome Specialty Dermatology Purple glove syndrome (PGS) is a poorly understood skin disease in which the extremities become swollen , discoloured and painful. [1] PGS is potentially serious, and may require amputation . PGS is most common among elderly patients and those receiving multiple large intravenous doses of the epilepsy drug phenytoin . [2] Compartment syndrome is a complication of PGS. Contents 1 Cause 2 Diagnosis 3 Treatment 4 References 5 External links Cause [ edit ] Purple glove syndrome is caused by the intravenous anticonvulsant phenytoin. ... Oral phenytoin can also result in development of purple glove syndrome. [3] References [ edit ] ^ Chokshi R, Openshaw J, Mehta NN, Mohler E (February 2007). "Purple glove syndrome following intravenous phenytoin administration". ... "Incidence and clinical consequence of the purple glove syndrome in patients receiving intravenous phenytoin".
-
H Syndrome
Wikipedia
Please improve this by adding secondary or tertiary sources . ( December 2018 ) ( Learn how and when to remove this template message ) H syndrome Other names Histiocytosis-lymphadenopathy plus syndrome This condition is inherited in an autosomal recessive manner H syndrome , also known as Histiocytosis-lymphadenopathy plus syndrome or PHID , [1] is a rare genetic condition caused by mutations in the SLC29A3 gene which encode the human equilibrative nucleoside transporter (hENT3) protein. [2] It is also known as Faisalabad histocytosis, familial Rosai-Dorfman disease, sinus histocytosis with massive lymphadenopathy and pigmented hypertrichosis with insulin-dependent diabetes mellitus syndrome. Contents 1 Presentation 2 Genetics 3 Pathogenesis 4 Management 5 History 6 References 7 External links Presentation [ edit ] This syndrome has a number of different clinical features many of which start with the letter 'H' giving rise to the name of the syndrome. ... ISBN 978-0-19-027648-5 . ^ Moynihan L M, Bundey SE, Heath D, Jones EL, McHale DP, Mueller RF, Markham, AF, Lench NJ (1998) Autozygosity mapping, to chromosome 11q25, of a rare autosomal recessive syndrome causing histiocytosis, joint contractures, and sensorineural deafness. ... Mutations in SLC29A3, encoding an equilibrative nucleoside transporter ENT3, cause a familial histiocytosis syndrome (Faisalabad histiocytosis) and familial Rosai-Dorfman disease. PLoS Genet. 6: e1000833 ^ Moynihan L M, Bundey SE, Heath D, Jones EL, McHale DP, Mueller RF, Markham, AF, Lench NJ (1998) Autozygosity mapping, to chromosome 11q25, of a rare autosomal recessive syndrome causing histiocytosis, joint contractures, and sensorineural deafness.

-
Riddle Syndrome
Wikipedia
Please improve this by adding secondary or tertiary sources . ( December 2018 ) ( Learn how and when to remove this template message ) RIDDLE syndrome Other names Radiosensitivity-immunodeficiency-dysmorphic features-learning difficulties syndrome Riddle syndrome is inherited in an autosomal recessive pattern. RIDDLE syndrome is a rare genetic syndrome. The name is an acronym for Radiosensitivity, ImmunoDeficiency Dysmorphic features and LEarning difficulties. ... Only four cases have been described up to 2017. [2] History [ edit ] This syndrome was first described by Stewart et al. 2007. [3] References [ edit ] ^ Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, et al. (February 2009). "The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage". ... (October 2007). "RIDDLE immunodeficiency syndrome is linked to defects in 53BP1-mediated DNA damage signaling" .

-
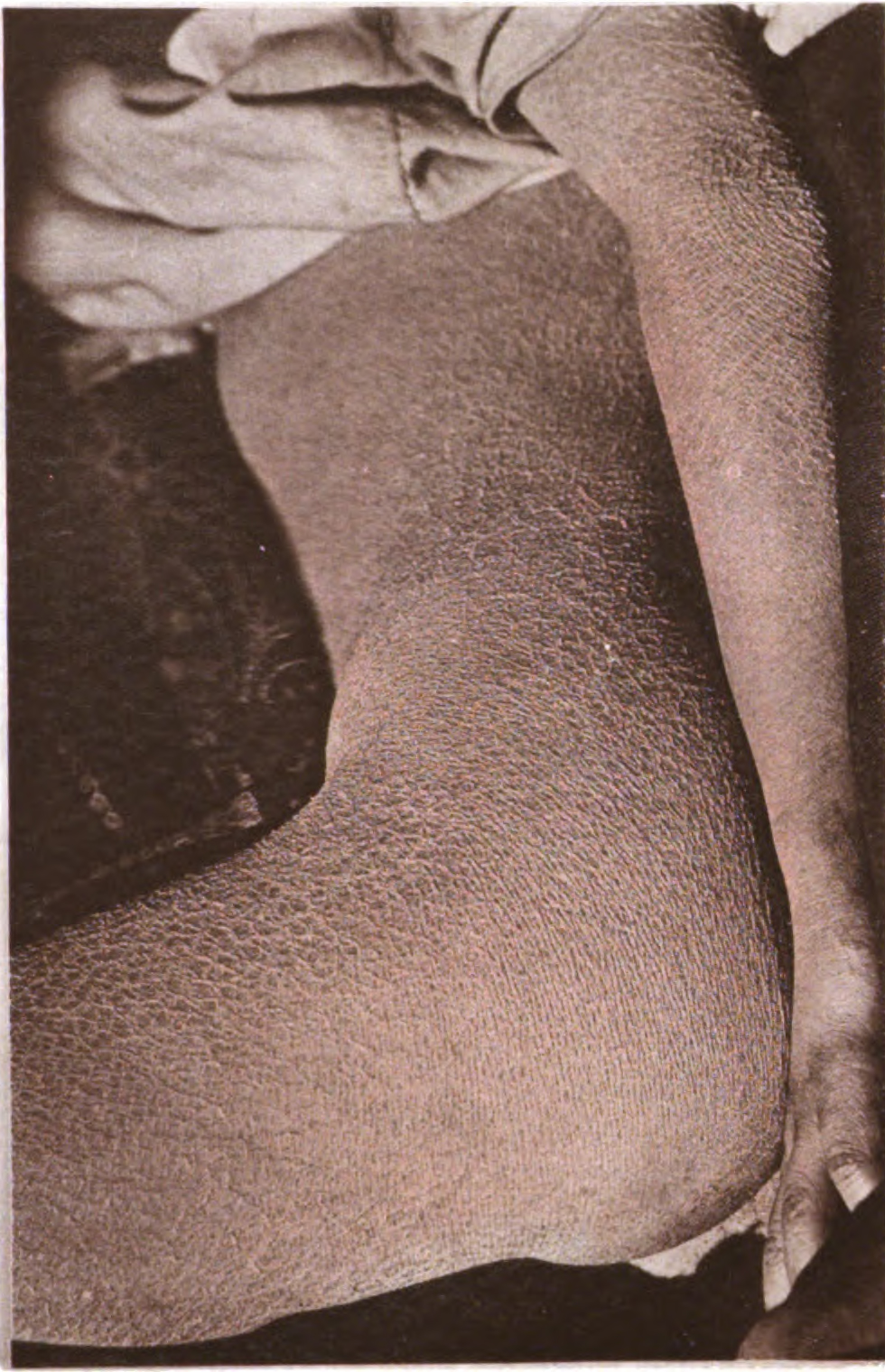
Ruzicka Goerz Anton Syndrome
Wikipedia
Ruzicka Goerz Anton syndrome Patient with Ichthyosis Ruzicka Goerz Anton syndrome is a rare genetic disease described by Ruzicka et al. in 1981. ... One year earlier, the patient had developed thyroid carcinoma , but whether or not this is due to the syndrome is unknown. The patient was treated with an oral retinoid, which greatly improved the patient's ichthyosis. [4] See also [ edit ] Ichthyosis Brachydactyly Clinodactyly References [ edit ] ^ a b "Ruzicka Goerz Anton syndrome" . ... Retrieved 2011-09-02 . ^ "Ruzicka-Goerz-Anton syndrome" (in Finnish). Mental Retardation Service Database. ... Retrieved 2011-09-01 . ^ "Ruzicka Goerz Anton syndrome" . National Library of Medicine. 2010-08-25 . Retrieved 2011-09-02 . ^ a b Ruzicka T, Goerz G, Anton-Lamprecht I (1981). "Syndrome of ichthyosis congenita, neurosensory deafness, oligophrenia, dental aplasia, brachydactyly, clinodactyly, accessory cervical ribs and carcinoma of the thyroid".
-
Pacs1-Related Syndrome
Gard
PACS-1 related syndrome causes intellectual disability, developmental delay, and distinctive facial features. This condition is present from birth. Symptoms of PACS-1 related syndrome may include low muscle tone , feeding difficulties, constipation, seizures, and heart defects. PACS1-related syndrome is caused by a specific genetic variant in the PACS1 gene. ... As of 2017, less than 70 people have been diagnosed with this syndrome. Treatment for PACS1-related syndrome is focused on managing the symptoms and may include medications to prevent seizures and placement of a feeding tube to help with weight gain.
-
Ross' Syndrome
Wikipedia
Symptoms of Adie syndrome plus segmental anhidrosis Ross' syndrome Specialty Dermatology Ross' syndrome consists of Adie's syndrome (myotonic pupils and absent deep tendon reflexes) plus segmental anhidrosis (typically associated with compensatory hyperhidrosis ). [1] It was characterized in 1958 [2] [3] by A.T. ... Retrieved 20 November 2010 . ^ Yaşar S, Aslan C, Serdar ZA, Demirci GT, Tutkavul K, Babalik D (April 2010). "Ross syndrome: Unilateral hyperhidrosis, Adie's tonic pupils and diffuse areflexia". ... "Progressive selective sudomotor denervation; a case with coexisting Adie's syndrome". Neurology . 8 (11): 809–17. doi : 10.1212/wnl.8.11.809 . ... "Tonic pupil, areflexia, and segmental anhidrosis: two additional cases of Ross syndrome and review of the literature". ... External links [ edit ] Classification D ICD - 10 : L74.8 ( ILDS L74.840) v t e Disorders of skin appendages Nail thickness: Onychogryphosis Onychauxis color: Beau's lines Yellow nail syndrome Leukonychia Azure lunula shape: Koilonychia Nail clubbing behavior: Onychotillomania Onychophagia other: Ingrown nail Anonychia ungrouped: Paronychia Acute Chronic Chevron nail Congenital onychodysplasia of the index fingers Green nails Half and half nails Hangnail Hapalonychia Hook nail Ingrown nail Lichen planus of the nails Longitudinal erythronychia Malalignment of the nail plate Median nail dystrophy Mees' lines Melanonychia Muehrcke's lines Nail–patella syndrome Onychoatrophy Onycholysis Onychomadesis Onychomatricoma Onychomycosis Onychophosis Onychoptosis defluvium Onychorrhexis Onychoschizia Platonychia Pincer nails Plummer's nail Psoriatic nails Pterygium inversum unguis Pterygium unguis Purpura of the nail bed Racquet nail Red lunulae Shell nail syndrome Splinter hemorrhage Spotted lunulae Staining of the nail plate Stippled nails Subungual hematoma Terry's nails Twenty-nail dystrophy Hair Hair loss / Baldness noncicatricial alopecia : Alopecia areata totalis universalis Ophiasis Androgenic alopecia (male-pattern baldness) Hypotrichosis Telogen effluvium Traction alopecia Lichen planopilaris Trichorrhexis nodosa Alopecia neoplastica Anagen effluvium Alopecia mucinosa cicatricial alopecia : Pseudopelade of Brocq Central centrifugal cicatricial alopecia Pressure alopecia Traumatic alopecia Tumor alopecia Hot comb alopecia Perifolliculitis capitis abscedens et suffodiens Graham-Little syndrome Folliculitis decalvans ungrouped: Triangular alopecia Frontal fibrosing alopecia Marie Unna hereditary hypotrichosis Hypertrichosis Hirsutism Acquired localised generalised patterned Congenital generalised localised X-linked Prepubertal Acneiform eruption Acne Acne vulgaris Acne conglobata Acne miliaris necrotica Tropical acne Infantile acne / Neonatal acne Excoriated acne Acne fulminans Acne medicamentosa (e.g., steroid acne ) Halogen acne Iododerma Bromoderma Chloracne Oil acne Tar acne Acne cosmetica Occupational acne Acne aestivalis Acne keloidalis nuchae Acne mechanica Acne with facial edema Pomade acne Acne necrotica Blackhead Lupus miliaris disseminatus faciei Rosacea Perioral dermatitis Granulomatous perioral dermatitis Phymatous rosacea Rhinophyma Blepharophyma Gnathophyma Metophyma Otophyma Papulopustular rosacea Lupoid rosacea Erythrotelangiectatic rosacea Glandular rosacea Gram-negative rosacea Steroid rosacea Ocular rosacea Persistent edema of rosacea Rosacea conglobata variants Periorificial dermatitis Pyoderma faciale Ungrouped Granulomatous facial dermatitis Idiopathic facial aseptic granuloma Periorbital dermatitis SAPHO syndrome Follicular cysts " Sebaceous cyst " Epidermoid cyst Trichilemmal cyst Steatocystoma simplex multiplex Milia Inflammation Folliculitis Folliculitis nares perforans Tufted folliculitis Pseudofolliculitis barbae Hidradenitis Hidradenitis suppurativa Recurrent palmoplantar hidradenitis Neutrophilic eccrine hidradenitis Ungrouped Acrokeratosis paraneoplastica of Bazex Acroosteolysis Bubble hair deformity Disseminate and recurrent infundibulofolliculitis Erosive pustular dermatitis of the scalp Erythromelanosis follicularis faciei et colli Hair casts Hair follicle nevus Intermittent hair–follicle dystrophy Keratosis pilaris atropicans Kinking hair Koenen's tumor Lichen planopilaris Lichen spinulosus Loose anagen syndrome Menkes kinky hair syndrome Monilethrix Parakeratosis pustulosa Pili ( Pili annulati Pili bifurcati Pili multigemini Pili pseudoannulati Pili torti ) Pityriasis amiantacea Plica neuropathica Poliosis Rubinstein–Taybi syndrome Setleis syndrome Traumatic anserine folliculosis Trichomegaly Trichomycosis axillaris Trichorrhexis ( Trichorrhexis invaginata Trichorrhexis nodosa ) Trichostasis spinulosa Uncombable hair syndrome Wooly hair nevus Sweat glands Eccrine Miliaria Colloid milium Miliaria crystalline Miliaria profunda Miliaria pustulosa Miliaria rubra Occlusion miliaria Postmiliarial hypohidrosis Granulosis rubra nasi Ross’ syndrome Anhidrosis Hyperhidrosis Generalized Gustatory Palmoplantar Apocrine Body odor Chromhidrosis Fox–Fordyce disease Sebaceous Sebaceous hyperplasia This condition of the skin appendages article is a stub .
-
Bare Lymphocyte Syndrome
Wikipedia
Bare lymphocyte syndrome Specialty Hematology Bare lymphocyte syndrome is a condition caused by mutations in certain genes of the major histocompatibility complex or involved with the processing and presentation of MHC molecules. ... As a notable contrast, however, bare lymphocyte syndrome does not result in decreased B- and T-cell counts, as the development of these cells is not impaired. ... "Associations and interactions between bare lymphocyte syndrome factors" . Mol. Cell. Biol . 20 (17): 6587–99. doi : 10.1128/MCB.20.17.6587-6599.2000 . ... Archived from the original on 2007-02-17. ^ Reith W, Mach B (2001). "The bare lymphocyte syndrome and the regulation of MHC expression". ... PMID 10417269 . ^ Online Mendelian Inheritance in Man (OMIM): 604571 External links [ edit ] Classification D ICD - 10 : D81.6 OMIM : 604571 209920 MeSH : D016511 DiseasesDB : 29570 v t e Lymphoid and complement disorders causing immunodeficiency Primary Antibody / humoral ( B ) Hypogammaglobulinemia X-linked agammaglobulinemia Transient hypogammaglobulinemia of infancy Dysgammaglobulinemia IgA deficiency IgG deficiency IgM deficiency Hyper IgM syndrome ( 1 2 3 4 5 ) Wiskott–Aldrich syndrome Hyper-IgE syndrome Other Common variable immunodeficiency ICF syndrome T cell deficiency ( T ) thymic hypoplasia : hypoparathyroid ( Di George's syndrome ) euparathyroid ( Nezelof syndrome Ataxia–telangiectasia ) peripheral: Purine nucleoside phosphorylase deficiency Hyper IgM syndrome ( 1 ) Severe combined (B+T) x-linked: X-SCID autosomal: Adenosine deaminase deficiency Omenn syndrome ZAP70 deficiency Bare lymphocyte syndrome Acquired HIV/AIDS Leukopenia : Lymphocytopenia Idiopathic CD4+ lymphocytopenia Complement deficiency C1-inhibitor ( Angioedema / Hereditary angioedema ) Complement 2 deficiency / Complement 4 deficiency MBL deficiency Properdin deficiency Complement 3 deficiency Terminal complement pathway deficiency Paroxysmal nocturnal hemoglobinuria Complement receptor deficiencyAK2, CD3D, CD3E, JAK3, ZAP70, TFRC, CIITA, HLA-C, B2M, F10, RFXANK, RFXAP, TAP1, H3C9P, ANKRA2, NT5C3A, MAPK1, RFX5, RFX1, ANXA2, MNAT1, IL2, HLA-A, H3P29
-
Sticky Platelet Syndrome
Wikipedia
Sticky platelet syndrome Specialty Hematology Sticky platelet syndrome is a term used by some [1] [2] [3] [4] to describe a disorder of platelet function. [5] It was first described by Mammen in 1983. [6] It is inherited in an autosomal dominant pattern. [7] It has not been associated with a specific gene, and it is not recognized as an entity in OMIM . ... PMID 10548069 . ^ Frenkel EP, Mammen EF (February 2003). "Sticky platelet syndrome and thrombocythemia". Hematol. ... "Bilateral Simultaneous Anterior Ischaemic Optic Neuropathy Associated with Sticky Platelet Syndrome". Br J Ophthalmol . 93 (7): 885–6, 913. doi : 10.1136/bjo.2008.142919 . ... "Protein S deficiency, activated protein C resistance and sticky platelet syndrome in a young woman with bilateral strokes" . ... PMID 12565719 . ^ Mammen EF (1999). "Sticky platelet syndrome" . Semin. Thromb. Hemost . 25 (4): 361–5. doi : 10.1055/s-2007-994939 .
-
Yentl Syndrome
Wikipedia
The Yentl Syndrome is the different course of action that heart attacks usually follow for women than for men. ... Bernadine Healy titled "The Yentl syndrome." References [ edit ] Bairey Merz, C. Noel (August 2011). "The Yentl syndrome and gender inequality in ischemic HD" . ... Orth-Gomer, Kristina (2000). "New light on the Yentl syndrome" (PDF) . European Heart Journal . 21 (11): 874–875. doi : 10.1053/euhj.1999.2025 . ... Thomas, Carolyn (17 April 2013), Yentl Syndrome: cardiology's gender gap is alive and well , Heart Sisters , retrieved 25 November 2014 Yentl's syndrome , Whonamedit?
-
Asperger Syndrome, Susceptibility To, 3
Omim
Description Asperger syndrome is considered to be a form of childhood autism (see, e.g., 209850). The DSM-IV (American Psychiatric Association, 1994) specifies several diagnostic criteria for Asperger syndrome, which has many of the same features as autism. In general, patients with Asperger syndrome and autism exhibit qualitative impairment in social interaction, as manifest by impairment in the use of nonverbal behaviors such as eye-to-eye gaze, facial expression, body postures, and gestures, failure to develop appropriate peer relationships, and lack of social sharing or reciprocity. ... Gillberg et al. (2001) described the development of the Asperger syndrome (and high-functioning autism) Diagnostic Interview (ASDI), which they claimed has a strong validity in the diagnosis of the disorder. For a discussion of genetic heterogeneity of Asperger syndrome, see ASPG1 (608638). Mapping Ylisaukko-oja et al. (2004) performed a genomewide scan on 17 Finnish families ascertained for Asperger syndrome with a strictly defined phenotype.
-
Sick Sinus Syndrome 3, Susceptibility To
Omim
A number sign (#) is used with this entry because of evidence that susceptibility to sick sinus syndrome-3 (SSS3) results from variation in the MYH6 gene (160710) on chromosome 14q11. Description Sick sinus syndrome may be encountered at any age but is primarily a disease of the elderly and is often secondary to other cardiac disorders when diagnosed in younger individuals. ... For a general phenotypic description and a discussion of genetic heterogeneity of sick sinus syndrome, see SSS1 (608567). Molecular Genetics Through complementary application of SNP genotyping, whole genome sequencing, and imputation in 38,384 Icelanders, Holm et al. (2011) identified MYH6 as a previously unidentified sick sinus syndrome susceptibility gene. A missense variant in this gene (160710.0008), arg721 to trp (2161C-T), has an allelic frequency of 0.38% in Icelanders and associates with sick sinus syndrome with an odds ratio of 12.53 (p = 1.5 x 10(-29)). Holm et al. (2011) showed that the lifetime risk of being diagnosed with sick sinus syndrome is around 6% for noncarriers of this variant but is approximately 50% for carriers of the variant.MYH6, SCN5A, HCN4, POMC, CACNA1D, KCNJ5, HCN1, SCN10A, CACNA1C, KCNE5, GPD1L, GNB5, AKAP9, PKP2, RANGRF, KCNE3, SLMAP, TRPM4, SCN3B, SCN1B, SGO1, ABCC9, SCN2B, CACNA2D1, KCNJ8, CACNB2, CALM2, KCND3, MECP2, PDCD1, ICAM1, TRPM7, COL1A2, TXN2, CCN2, FBN1, FGFR3, FN1, IL6, IGF1, MEF2C, HDAC4, TBX18, VEGFA, VCAM1, TTN, THBS1, TGFB1, CCL2, LMNA, AGT
-
Ivic Syndrome
Orphanet
IVIC syndrome is a very rare genetic malformation syndrome characterized by upper limb anomalies (radial ray defects, carpal bone fusion), extraocular motor disturbances, and congenital bilateral non-progressive mixed hearing loss. ... There have been reports of sudden death. Etiology The syndrome has been linked to mutations in the SALL4 gene (20q13.2) encoding a transcription factor involved in the maintenance and self-renewal of embryonic and hematopoietic stem cells. Okihiro syndrome (see this term) is a disorder allelic to IVIC syndrome. Genetic counseling IVIC syndrome is inherited in an autosomal dominant manner. Genetic counseling should be offered to affected families, informing them of the 50% risk of offspring inheriting the disease-causing mutation and therefore being affected with the syndrome.
-
Nablus Mask-Like Facial Syndrome
Wikipedia
Nablus mask-like facial syndrome Other names 8q22.1 microdeletion syndrome Nablus mask-like facial syndrome (Nablus syndrome) is a rare (13 cases described by 2018) genetic condition. It is a microdeletion syndrome triggered by a deletion at chromosome 8 q22.1 that causes a mask-like facial appearance in those affected. [1] Contents 1 Presentation 2 Genetics 3 Epidemiology 4 References 5 External links Presentation [ edit ] It is characterized by a narrowing of the eyes , tight, glistening facial skin, and a flat, broad nose. Other features of the syndrome include malformed ears, unusual hair patterns on the scalp, bent fingers and toes and joint deformities in the hands and feet, unusual teeth, mild developmental delay , cryptorchidism . Genetics [ edit ] This syndrome is associated with a deletion mutation in the long arm of chromosome 8 (8q22.1). [ citation needed ] Its inheritance pattern has not yet been determined. [ citation needed ] Epidemiology [ edit ] It is a rare genetic disorder by inheritance found in Palestine in 2000 by Ahmad Teebi.
-
Cornelia De Lange Syndrome
Medlineplus
Cornelia de Lange syndrome is a developmental disorder that affects many parts of the body. ... Frequency Although the exact incidence is unknown, Cornelia de Lange syndrome likely affects 1 in 10,000 to 30,000 newborns. The condition is probably underdiagnosed because affected individuals with mild or uncommon features may never be recognized as having Cornelia de Lange syndrome. Causes Cornelia de Lange syndrome can result from mutations in at least five genes: NIPBL , SMC1A , HDAC8 , RAD21 , and SMC3 . ... Learn more about the genes associated with Cornelia de Lange syndrome HDAC8 NIPBL RAD21 SMC1A SMC3 Inheritance Pattern When Cornelia de Lange syndrome is caused by mutations in the NIPBL , RAD21 , or SMC3 gene, the condition is considered to have an autosomal dominant pattern of inheritance. ... Studies of X-linked Cornelia de Lange syndrome indicate that one copy of the altered gene in each cell may be sufficient to cause the condition.
-
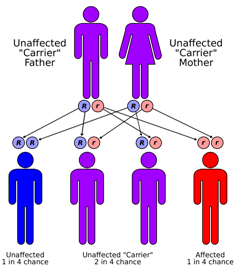
Sanjad-Sakati Syndrome
Wikipedia
Sanjad-Sakati syndrome Other names Hypoparathyroidism-short stature-intellectual disability-seizures syndrome Sanjad-Sakati syndrome is inherited in an autosomal recessive manner Usual onset diagnosis is clinically by features of triad of growth retardation, dysmorphic features and hypo calcemic tetany or sezures Sanjad-Sakati syndrome is a rare autosomal recessive genetic condition seen in offspring of Middle Eastern origin. ... Typically, children with this syndrome are born low-birth-weight due to intrauterine growth retardation. ... "Congenital hypoparathyroidism with dysmorphic features: a new syndrome. (Abstract)". Pediatric Research . 23 : 271A. ^ Sanjad, S. ... K.; Kaddoura, R.; Milner, R. D. (February 1991). "A new syndrome of congenital hypoparathyroidism, severe growth failure, and dysmorphic features" . ... Hypoparathyroidism-retardation-dysmorphism syndrome in a girl: a new variant not caused by a TBCE mutation--clinical report and review.
-
Pseudo-Cushing's Syndrome
Wikipedia
Pseudo-Cushing's syndrome Specialty Endocrinology Pseudo-Cushing's syndrome is a medical condition in which patients display the signs , symptoms , and abnormal hormone levels seen in Cushing's syndrome . ... Estrogen can cause an increase of cortisol-binding globulin and thereby cause the total cortisol level to be elevated. [2] Contents 1 Diagnosis 1.1 Differential diagnosis 2 Prognosis 3 References 4 External links Diagnosis [ edit ] Levels of cortisol and ACTH both elevated 24-hour urinary cortisol levels elevated Dexamethasone suppression test [3] Late night salivary cortisol (LNSC) [4] Loss of diurnal variation in cortisol levels (seen only in true Cushing's Syndrome) High mean corpuscular volume and gamma-glutamyl transferase may be clues to alcoholism Polycystic Ovarian Syndrome should be ruled out; PCOS may have similar symptoms Differential diagnosis [ edit ] Differentiation from Cushing's is difficult, but several tools exist to aid in the diagnosis [5] Alternative causes of Cushing's should be excluded with imaging of lungs , adrenal glands , and pituitary gland ; these often appear normal in Cushing's In the alcoholic patient with pseudo-Cushing's, admission to hospital (and avoidance of alcohol) will result in normal midnight cortisol levels within five days, excluding Cushing's [6] Another cause for Cushing's syndrome is adrenocortical carcinoma . ... "Reevaluation of the combined dexamethasone suppression-corticotropin-releasing hormone test for differentiation of mild cushing's disease from pseudo-Cushing's syndrome" . Journal of Clinical Endocrinology and Metabolism . 92 (11): 4290–3. doi : 10.1210/jc.2006-2829 . ... "The diagnosis and differential diagnosis of Cushing's syndrome and pseudo-Cushing's states". Endocrine Reviews . 19 (5): 647–72. doi : 10.1210/er.19.5.647 . ... External links [ edit ] Classification D ICD - 10 : E24.3 External resources eMedicine : med/1936 v t e Adrenal gland disorder Hyperfunction Aldosterone Hyperaldosteronism Primary aldosteronism Conn syndrome Bartter syndrome Glucocorticoid remediable aldosteronism AME Liddle's syndrome 17α CAH Pseudohypoaldosteronism Cortisol Cushing's syndrome Pseudo-Cushing's syndrome Steroid-induced osteoporosis Sex hormones 21α CAH 11β CAH Hypofunction Aldosterone Hypoaldosteronism 21α CAH 11β CAH Cortisol CAH Lipoid 3β 11β 17α 21α Sex hormones 17α CAH Inborn errors of steroid metabolism Adrenal insufficiency Adrenal crisis Adrenalitis Xanthogranulomatous Addison's disease Waterhouse–Friderichsen syndrome




