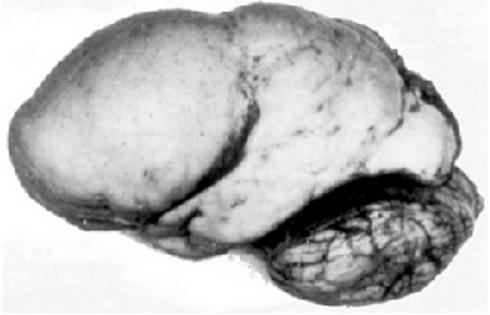
Hardikar syndrome is a very rare multiple congenital malformation syndrome characterized by obstructive liver and kidney disease, intestinal malrotation , genitourinary abnormalities, cleft lip and palate , pigmentary retinopathy (breakdown of the light-sensing tissue at the back of the eye), and congenital heart defects. ... The cause of this condition remains unknown, although an overlap with Kabuki syndrome and Alagille syndrome have been debated.
Nydegger et al. (2008) noted that the syndrome resembled the Kabuki (147920) and Alagille (ALGS1; 118450) syndromes. Cools and Jaeken (1997) reported an infant girl with Hardikar syndrome. She had cleft lip and palate, pigmentary retinopathy, hydroureteronephrosis with severe vesicoureteral reflux, cholestasis, and nonrotation of the gut. ... Cools and Jaeken (1997) had noted that the syndrome resembled the Kabuki syndrome. Maluf et al. (2002) reported a girl with features consistent with Hardikar syndrome. She had cleft lip and palate, retinopathy, intestinal malrotation, and recurrent urinary tract infections associated with stenosis of the ureters. ... Poley and Proud (2008) reported another girl with Hardikar syndrome who had malrotation of the gut, pigmentary retinopathy, patent ductus arteriosus, hydronephrosis, and persistent cholestasis resulting in liver failure and transplant.
Cholestasis-pigmentary retinopathy-cleft palate is a syndrome of multiple congenital malformations, characterized by an association of cleft lip and palate, patchy pigmentary retinopathy (cat's paw), obstructive liver disease (cholestasis, portal hypertension etc.) and obstructive renal disease (ectopic ureteric insertion, obstruction, vesicouretral reflux and hydronephrosis). Gastrointestinal tract involvement (malrotation, gastresophageal reflux etc.) and cardiac involvement (coarctation of aorta, pulmonary artery stenosis, etc.) have also been reported. An overlap with Kabuki syndrome is debated.
MORM syndrome Other names Intellectual disability-truncal obesity-retinal dystrophy-micropenis syndrome, Mental retardation-truncal obesity-retinal dystrophy-micropenis syndrome The image shows chromosome 9. ... MORM syndrome is an autosomal recessive congenital disorder [1] characterized by mental retardation , truncal obesity, retinal dystrophy , and micropenis ". [1] The disorder shares similar characteristics with Bardet–Biedl syndrome and Cohen syndrome , both of which are autosomal recessive genetic disorders. [1] [2] MORM syndrome can be distinguished from the above disorders because symptoms appear at a young age. [1] The disorder is not dependent on sex of the offspring, both male and female offspring are equally likely to inherit the disorder. The syndrome is caused by a mutation in the INPP5E gene which can be located on chromosome 9 in humans. [3] Further mapping resulted in the identification of a MORM syndrome locus on chromosome 9q34.3 between the genetic markers D9S158 and D9S905. [4] Contents 1 Presentation 2 Genetic 3 Diagnosis 4 Management 5 References 6 External links Presentation [ edit ] For individuals with MORM syndrome, symptoms do not appear until about one year of age. [1] From conception to birth, individuals with MORM syndrome appear asymptotic, with no abnormal characteristics. [1] Vision is negatively affected within the first year of life, particularly night vision. [1] Individuals with MORM syndrome experience decreased visual acuity , meaning their ability to see distinct sharp lines decreases. [1] [5] Vision quality continues to deteriorate until age three. [1] Any further reduction in vision acuity is not observed until the individual is between the ages thirty to forty. [1] Delayed sentence processing and intellectual disability is associated with individuals with MORM syndrome, primarily observed at age four. [1] Individuals continue to develop and grow until they are five to twelve years old. ... H.; Woods, C. G. (May 2006). "MORM syndrome (mental retardation, truncal obesity, retinal dystrophy and micropenis), a new autosomal recessive disorder, links to 9q34" (PDF) . ... Retrieved 3 December 2015 . ^ a b c "MORM syndrome" . Genetic Testing Registry . National Centre for Biotechnological Information .
A number sign (#) is used with this entry because of evidence that a syndrome of mental retardation, truncal obesity, retinal dystrophy, and micropenis syndrome (MORMS) is caused by homozygous mutation in the INPP5E gene (613037) on chromosome 9q34. ... The authors suggested the acronym 'MORM' syndrome. The phenotype was similar to Bardet-Biedl syndrome (BBS; 209900) and Cohen syndrome (COH1; 216550) but could be distinguished by the age of onset and nonprogressive nature of the visual impairment, and the lack of several characteristics, including dysmorphic facies, skin or gingival infection, microcephaly, 'mottled retina,' polydactyly, and testicular anomalies. ... Mapping By genomewide linkage analysis, Hampshire et al. (2006) identified a candidate MORM syndrome locus within an approximately 1-cM subtelomeric region on chromosome 9q34.3 (maximum lod score of 5.64) between markers D9S158 and D9S905. Molecular Genetics In affected members of a family with MORM syndrome, Jacoby et al. (2009) identified a homozygous mutation (Q627X; 613037.0001) in the INPP5E gene.
A rare genetic syndromic intellectual disability characterized by language delay and mild to moderate intellectual disability associated with truncal obesity, congenital nonprogressive retinal dystrophy with poor night vision and reduced visual acuity, and micropenis in males.
Psychological syndrome where men resist aging to an unusual degree Dorian Gray syndrome ( DGS ) denotes a cultural and societal phenomenon characterized by a man's extreme pride in his personal appearance and the fitness of his physique, which is accompanied by difficulties in coping with the requirements of psychological maturation and with the aging of his body. ... The personal character of the man Dorian Gray is the background for the clinical description of the Dorian Gray syndrome that afflicts the patient. Causes [ edit ] The Dorian Gray syndrome arises from the concurring and overlapping clinical concepts of the narcissistic personality , dysmorphophobia , and paraphilia . ... Further reading [ edit ] Brosig B.(2000) The "Dorian Gray Syndrome" and other fountains of youth. ... Brosig, B., Euler, S., Brähler, E., Gieler, U. (2005) Das Dorian Gray Syndrom. In: Trüeb, R. A. (Hg.) : Smart aging. ... Euler, S., Brähler, E., Brosig, B. (2003): Das Dorian-Gray-Syndrom als „ethnische Störung“ der Spätmoderne.
Fetal alcohol syndrome causes brain damage and growth problems. The problems caused by fetal alcohol syndrome vary from child to child, but defects caused by fetal alcohol syndrome are not reversible. ... If you drink during pregnancy, you place your baby at risk of fetal alcohol syndrome. If you suspect your child has fetal alcohol syndrome, talk to your doctor as soon as possible. ... Coping and support The psychological and emotional problems associated with fetal alcohol syndrome can be difficult to manage for the person with the syndrome and for the family. Family support Children with fetal alcohol syndrome and their families may benefit from the support of professionals and other families who have experience with this syndrome.
Outdated medical term Gay bowel syndrome is a term that was first used by Henry L. ... Sometimes, difficulty in specifying the method may be a result of transmission by both methods. [3] Following the onset of the AIDS epidemic , the reported incidence of these complaints has declined, likely as a result of safer sexual practices . [4] Those with the ano-rectal disorder experience increased incidents of diarrhea . [5] Criticism and decline in use [ edit ] In 1985, an article in the peer-reviewed journal Gut said that gay bowel syndrome was not a syndrome, and had limitations in medical use: The "gay bowel syndrome" was first used to describe not a syndrome, but a list of conditions. ... "The gay bowel syndrome: clinico-pathologic correlation in 260 cases". ... PMID 9328857 . ^ a b Garbo, Jon (December 21, 2004). " " Gay Bowel Syndrome" struck from textbook" . Gmax.co.za . ... "Activist fights 'outdated' medical phrase: Effort to debunk 'gay bowel syndrome' may face new challenge" . Washington Blade .
Before identification of the gene in which mutation is causative, MYH9 , individuals with MYH9RD were diagnosed as having Epstein syndrome, Fechtner syndrome, May-Hegglin anomaly, or Sebastian syndrome based on the combination of different clinical findings at the time of diagnosis. ... Nomenclature In the past, the conditions included in MYH9RD were known as Epstein syndrome Fechtner syndrome May-Hegglin anomaly Sebastian syndrome (Sebastian platelet syndrome) These four disorders, characterized by thrombocytopenia with giant platelets, were classified on the basis of morphologic aspects of Döhle-like bodies and different combinations of the other manifestations of MYH9RD: hearing loss, glomerular nephropathy, and cataract. ... The hallmark of Gray platelet syndrome is the finding of "pale" platelets on peripheral blood films as a result of lack of alpha granules. ... Collagen IV-related nephropathies , including X-linked and autosomal (dominant and recessive) forms of Alport syndrome and thin basement membrane nephropathy. ... Platelet defects have not been described in either Alport syndrome or thin basement membrane nephropathy.
MYH9 -related disorder was previously thought to be four separate disorders: May-Hegglin anomaly, Epstein syndrome, Fechtner syndrome, and Sebastian syndrome.
Parkinsonism and psychiatric changes are usually the earliest features of Perry syndrome. Signs of parkinsonism include unusually slow movements (bradykinesia), stiffness, and tremors. ... Hypoventilation is a later feature of Perry syndrome. Abnormally slow breathing most often occurs at night, causing affected individuals to wake up frequently. ... Suicide is another cause of death in this condition. Frequency Perry syndrome is very rare; about 50 affected individuals have been reported worldwide. Causes Perry syndrome results from mutations in the DCTN1 gene. ... A gradual loss of neurons in areas of the brain that regulate movement, emotion, and breathing underlies the signs and symptoms of Perry syndrome. Learn more about the gene associated with Perry syndrome DCTN1 Inheritance Pattern This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
A number sign (#) is used with this entry because Perry syndrome is caused by heterozygous mutation in the DCTN1 gene (601143) on chromosome 2p13. ... Newsway et al. (2010) reported a man with Perry syndrome who developed progressive symptoms in his mid-forties. ... All had parkinsonism, and 1 also had psychiatric symptoms and respiratory failure, leading to a diagnosis of Perry syndrome. Another had oculomotor abnormalities, consistent with PSP. Overall, the clinical diagnoses were Parkinson disease in 4 patients, Perry syndrome in 1, progressive supranuclear palsy in 2, and frontotemporal dementia in 2. ... Neuropathologic Features By neuropathologic examination of 8 affected individuals, Wider et al. (2010) demonstrated that Perry syndrome is a TDP43 (605078)-related proteinopathy.
Summary Clinical characteristics. Perry syndrome is characterized by parkinsonism, hypoventilation, depression, and weight loss. ... Each child of an individual with Perry syndrome has a 50% chance of inheriting the pathogenic variant. ... Establishing the Diagnosis The diagnosis of Perry syndrome can be established in a proband who has ALL of the above cardinal features. ... An individual of Japanese ancestry who showed symptoms reminiscent of Perry syndrome was found to have an MAPT pathogenic variant. ... Response to levodopa is usually absent, erratic, or transient in Perry syndrome [Tsuboi et al 2002, Wider & Wszolek 2008].
., France, Turkey and Japan. Clinical description Perry syndrome has a mean age of onset of 48 years (range 35-61) and presents with parkinsonism (akinetic-rigid and rather symmetric), psychiatric changes manifesting as depression, lethargy, withdrawal, apathy, and changes in character, as well as sleep difficulties. The usual duration of Perry syndrome is about 5 years, with severe weight loss and central hypoventilation being seen late in the disease course. ... Patients are often bedridden or wheelchair bound as motor impairment may be severe at a later stage of the disease. Etiology Perry syndrome is caused by mutations (five identified to date) in exon 2 of the dynactin DCTN1 gene coding for p150glued, the major subunit of the dynactin protein complex. ... Management and treatment There is no cure for Perry syndrome. Symptomatic treatment requires a multidisciplinary team. ... With worsening symptoms hospitalization and major medical assistance is often required. Prognosis Perry syndrome progresses rapidly and the prognosis is poor.
Cobb syndrome is defined by the association of vascular cutaneous (venous or arteriovenous), muscular (arteriovenous), osseous (arteriovenous) and medullary (arteriovenous) lesions at the same metamere or spinal segment. ... Epidemiology Less than 100 cases of Cobb syndrome have been reported in the literature. There is no sex predilection. Cobb syndrome represents less than 15% of cases of spinal cord arteriovenous malformations. ... The cutaneous manifestations of the syndrome are most often flat vascular lesions (port-wine stains) but angiokeratomas, angiolipomas and lymphangiomas have been reported. ... Two consecutive territories may be affected resulting in multimetameric forms of the disease. Recent analysis of Cobb syndrome has led to use of the term Spinal Arteriovenous Metameric Syndrome 1-31 (SAMS 1-31), by analogy with the Cerebrofacial Arteriovenous Metameric Syndromes (CAMS 1-3) and the Cerebrofacial venous metameric syndromes (CVMS1-3).
Cobb syndrome Other names Spinal arteriovenous metameric syndrome Cobb syndrome is a rare congenital disorder characterized by visible skin lesions with underlying spinal angiomas or arteriovenous malformations (AVMs). [1] The skin lesions of Cobb syndrome typically are present as port wine stains or angiomas, but reports exist of angiokeratomas , [2] angiolipomas , and lymphangioma circumscriptum . [3] The intraspinal lesions may be angiomas or AVMs and occur at levels of the spinal cord corresponding to the affected skin dermatomes . ... A possible complication if treatment is delayed is Foix-Alajouanine syndrome [6] or subacute necrotic myelopathy due to thrombosis in the spinal angioma. ... ISBN 1-4160-2999-0 . ^ Clinton TS, Cooke LM, Graham BS (April 2003). "Cobb syndrome associated with a verrucous (angiokeratomalike) vascular malformation". ... PMID 12729091 . ^ Shim JH, Lee DW, Cho BK (1996). "A case of Cobb syndrome associated with lymphangioma circumscriptum". ... PMC 1406820 . PMID 17863459 . ^ a b "Cobb syndrome" . Genetic and Rare Diseases Information Center (GARD) – an NCATS Program .
Sulfonamide hypersensitivity syndrome Sulfonamide hypersensitivity syndrome is similar to anticonvulsant hypersensitivity syndrome , but the onset is often sooner in the treatment course, generally after 7–14 days of therapy. [1] : 118–9 It is considered immune-mediated. [2] See also [ edit ] List of cutaneous conditions References [ edit ] ^ James, William; Berger, Timothy; Elston, Dirk (2005). ... External links [ edit ] Classification D ICD - 10 : Y40 ICD - 9-CM : E930.8 v t e Adverse drug reactions Antibiotics Penicillin drug reaction Sulfonamide hypersensitivity syndrome Urticarial erythema multiforme Adverse effects of fluoroquinolones Red man syndrome Jarisch–Herxheimer reaction Hormones Steroid acne Steroid folliculitis Chemotherapy Chemotherapy-induced acral erythema Chemotherapy-induced hyperpigmentation Scleroderma-like reaction to taxanes Hydroxyurea dermopathy Exudative hyponychial dermatitis Anticoagulants Anticoagulant-induced skin necrosis Warfarin necrosis Vitamin K reaction Texier's disease Immunologics Adverse reaction to biologic agents Leukotriene receptor antagonist-associated Churg–Strauss syndrome Methotrexate-induced papular eruption Adverse reaction to cytokines Other drugs Anticonvulsant hypersensitivity syndrome Allopurinol hypersensitivity syndrome Vaccine adverse event Eczema vaccinatum Bromoderma Halogenoderma Iododerma General Skin and body membranes Acute generalized exanthematous pustulosis Bullous drug reaction Drug-induced acne Drug-induced angioedema Drug-related gingival hyperplasia Drug-induced lichenoid reaction Drug-induced lupus erythematosus Drug-induced nail changes Drug-induced pigmentation Drug-induced urticaria Stevens–Johnson syndrome Injection site reaction Linear IgA bullous dermatosis Toxic epidermal necrolysis HIV disease-related drug reaction Photosensitive drug reaction Other Drug-induced pseudolymphoma Fixed drug reaction Serum sickness-like reaction This cutaneous condition article is a stub .
Ackerman syndrome Other names Pyramidal molar-glaucoma-upper abnormal lip syndrome Ackerman syndrome is inherited in an autosomal recessive manner Ackerman syndrome is a familial syndrome of fused molar roots with a single canal ( taurodontism ), hypotrichosis , full upper lip without a cupid's bow, thickened and wide philtrum , and occasional juvenile glaucoma . [1] It was described by James L. ... "A New Dental, Ocular and Cutaneous Syndrome". International Journal of Dermatology . 12 (5): 285–89. doi : 10.1111/j.1365-4362.1973.tb00056.x . ... "Interstitial granulomatous dermatitis with arthritis (Ackerman syndrome)". J. Rheumatol . 33 (6): 1207–9. ... "Arthritis and interstitial granulomatous dermatitis (Ackerman syndrome) with pulmonary silicosis". Semin. ... PMID 12701044 . ^ "Symptoms of Ackerman syndrome - RightDiagnosis.com" . www.rightdiagnosis.com .
A rare multiple congenital anomalies/dysmorphic syndrome characterized by a variable combination of dental, cutaneous, ocular, and bone abnormalities, including pyramidal and fused molar roots, taurodontism, an abnormal upper lip without a cupid's bow and thickened and wide philtrum, juvenile glaucoma, syndactyly, and clinodactyly.
Clinical Features Ackerman et al. (1973) described a kindred in which a sister and 2 brothers had pyramidal molar roots. The 2 brothers had juvenile glaucoma and all 6 members of their sibship were said to have an unusual morphology of the upper lip which was full without a Cupid's bow and with a thickened and widened philtrum. Pyramidal, taurodont, or fused molar roots were found in all 3 otherwise unaffected sibs, in both parents, and in sibs, nephews, and nieces of both parents. Were these persons heterozygous for a gene that produced glaucoma and all pyramidal teeth in the 3 sibs who were homozygous? Mouth - Upper lip full - Cupid's bow absent - Philtrum thick and wide Inheritance - Autosomal recessive Eye - Glaucoma, juvenile Teeth - Molar roots pyramidal - Taurodontism ▲ Close
Troyer syndrome is part of a group of genetic disorders known as hereditary spastic paraplegias. ... People with Troyer syndrome can experience a variety of signs and symptoms. ... The severity of impairment related to Troyer syndrome increases as a person ages. Most affected individuals require a wheelchair by the time they are in their fifties or sixties. Frequency About 20 cases of Troyer syndrome have been reported in the Old Order Amish population of Ohio. It has not been found outside this population. Causes Troyer syndrome is caused by a mutation in the SPART gene.
Diagnosis/testing. The diagnosis of Troyer syndrome is established in an individual with characteristic clinical findings. ... Clinical Characteristics Clinical Description Troyer syndrome is characterized by both developmental and neurodegenerative processes. ... The cardinal features of Troyer syndrome include developmental delay, spastic paraparesis, dysarthria, distal amyotrophy, and short stature. ... A previous study documented 21 individuals with Troyer syndrome in a population of approximately 50,000 Amish [Patel et al 2002]. ... Some Amish children/infants have been diagnosed with Silver-Russell syndrome due to low birth weight, relatively preserved head circumference, triangular face shape, and short stature; however, spastic paraplegia, a consistent feature of Troyer syndrome, is not seen in individuals with Silver-Russell syndrome.
A number sign (#) is used with this entry because of evidence that autosomal recessive spastic paraplegia-20, also known as Troyer syndrome, is caused by homozygous mutation in the SPG20 gene, encoding spartin (607111), on chromosome 13q13. ... Clinical Features In an Amish group in Ohio, Cross and McKusick (1967) observed 20 cases of spastic paraplegia with distal muscle wasting, and designated it Troyer syndrome for the surname of many of the affected persons. ... Bakowska et al. (2008) reported an Amish brother and sister from Ohio with Troyer syndrome. Both showed delayed motor and cognitive development and developed a progressive deterioration in gait and speech during childhood. ... Mapping Patel et al. (2002) excluded 5 loci for autosomal recessive hereditary spastic paraplegia by linkage analysis and, using homozygosity mapping, mapped the Troyer syndrome gene to a 731-kb interval of chromosome 13q12.3. ... Molecular Genetics Patel et al. (2002) identified a frameshift mutation (1110delA; 607111.0001) in the SPG20 gene in individuals with Troyer syndrome from the Amish kindred in which the disorder was first described.
Troyer syndrome is a neurological disorder and one of the many types of hereditary spastic paraplegia . ... Life expectancy is normal. Troyer syndrome is caused by mutations in the SPG20 gene and is inherited in an autosomal recessive manner.
Autosomal recessive spastic paraplegia type 20 (SPG20) is a type of complex hereditary spastic paraplegia characterized by an onset in infancy of progressive spastic paraparesis associated with distal amyotrophy, psuedobulbar palsy, motor and cognitive delays, mild cerebellar signs (dysarthria, dysdiadochokinesia, mild intention tremor), short stature and subtle skeletal abnormalities (pes cavus, mild talipes equinovarus, kyphoscoliosis). SPG20 is due to mutations in the SPG20 gene (13q13.1), which encodes the protein spartin.
Acropectoral syndrome Acropectoral syndrome has an autosomal dominant pattern of inheritance. Acropectoral syndrome is an autosomal dominant skeletal dysplasia syndrome affecting the hands , feet, sternum , and lumbosacral spine . ... In some cases, this was accompanied by hypoplasia of the head of the first metatarsal and absence of both phalanges of the hallux . [1] Genetics [ edit ] The cytogenetic location is 7q36 and genomic coordinates are GRCh37:147,900,000 - 159,138,663 (NCBI). Mapping of this syndrome was done by Dundar and coworkers in 2001. ... Dundar and coworkers characterized and mapped acropectoral syndrome and also showed it was unrelated to acropectorovertebral syndrome. ... (May 2001). "A novel acropectoral syndrome maps to chromosome 7q36" . J. Med.
A rare syndrome characterized by a combination of distal limb abnormalities (syndactyly of all fingers and toes, preaxial polydactyly in the feet and/or hands) and upper sternum malformations.
Dundar et al. (2001) pointed out the similarity between this phenotype and acropectorovertebral dysplasia (F syndrome; 102510) but noted that in this syndrome the carpal, tarsal, and metatarsal synostoses and vertebral anomalies present in F syndrome were not seen. In addition, the soft tissue syndactyly was more marked than that seen in F syndrome, and the preaxial polydactyly occurred in the feet as well as in the hands. Prashanth et al. (2012) reported features suggestive of acropectoral syndrome in members of a nonconsanguineous family from southern India. ... INHERITANCE - Autosomal dominant SKELETAL Hands - Preaxial polydactyly - Bifid thumb - Triphalangeal thumb - Soft tissue syndactyly between all fingers (in 1 family) Feet - Soft tissue syndactyly between all toes (in 1 family) - Preaxial polydactyly NEUROLOGIC Central Nervous System - Mental retardation, moderate (in 1 patient) MISCELLANEOUS - Variable phenotype - F syndrome ( 102510 ) has many overlapping features - Two families reported (last curated September 2012) ▲ Close
Hypermobile joints are a feature of genetic connective tissue disorders such as hypermobility spectrum disorder (HSD) or Ehlers–Danlos syndromes . Until new diagnostic criteria were introduced, hypermobility syndrome was sometimes considered identical to Ehlers–Danlos syndrome hypermobile type/EDS Type 3. ... Hypermobility may be symptomatic of a serious medical condition, such as Stickler syndrome , Ehlers–Danlos syndrome , [8] Marfan syndrome , [8] Loeys–Dietz syndrome , rheumatoid arthritis , osteogenesis imperfecta , [8] lupus , polio , Down syndrome , [8] morquio syndrome , cleidocranial dysostosis or myotonia congenita . ... Diagnosis [ edit ] Joint hypermobility syndrome shares symptoms with other conditions such as Marfan syndrome, Ehlers-Danlos Syndrome, and osteogenesis imperfecta . ... Joint hypermobility handbook : a guide for the issues & management of Ehlers-Danlos syndrome hypermobility type and the hypermobility syndrome . ... "Tenascin-X: a candidate gene for benign joint hypermobility syndrome and hypermobility type Ehlers-Danlos syndrome?"
In heritable connective tissue disorders associated with joint hyper-mobility (such as Marfan syndrome and Ehlers–Danlos syndrome types I–III, VII, and XI), the joint laxity usually is apparent before adulthood. However, age of onset and extent of joint laxity are variable in Marfan syndrome, and joint laxity may be confined to the hands alone, as in Ehlers–Danlos syndrome type I. ... However, if there is widespread laxity of other connective tissue, then this may be a sign of Ehlers-Danlos syndrome. Ligamentous laxity may also result from injury, such as from a vehicle accident. ... External links [ edit ] Classification D ICD - 10 : M24.2 ICD - 9-CM : 728.4 v t e Acquired musculoskeletal deformities Upper limb shoulder Winged scapula Adhesive capsulitis Rotator cuff tear Subacromial bursitis elbow Cubitus valgus Cubitus varus hand deformity Wrist drop Boutonniere deformity Swan neck deformity Mallet finger Lower limb hip Protrusio acetabuli Coxa valga Coxa vara leg Unequal leg length patella Luxating patella Chondromalacia patellae Patella baja Patella alta foot deformity Bunion/hallux valgus Hallux varus Hallux rigidus Hammer toe Foot drop Flat feet Club foot knee Genu recurvatum Head Cauliflower ear General terms Valgus deformity / Varus deformity Joint stiffness Ligamentous laxity v t e Soft tissue disorders Capsular joint Synoviopathy Synovitis / Tenosynovitis Calcific tendinitis Stenosing tenosynovitis Trigger finger De Quervain syndrome Transient synovitis Ganglion cyst osteochondromatosis Synovial osteochondromatosis Plica syndrome villonodular synovitis Giant-cell tumor of the tendon sheath Bursopathy Bursitis Olecranon Prepatellar Trochanteric Subacromial Achilles Retrocalcaneal Ischial Iliopsoas Synovial cyst Baker's cyst Calcific bursitis Noncapsular joint Symptoms Ligamentous laxity Hypermobility Enthesopathy / Enthesitis / Tendinopathy upper limb Adhesive capsulitis of shoulder Impingement syndrome Rotator cuff tear Golfer's elbow Tennis elbow lower limb Iliotibial band syndrome Patellar tendinitis Achilles tendinitis Calcaneal spur Metatarsalgia Bone spur other/general: Tendinitis / Tendinosis Nonjoint Fasciopathy Fasciitis : Plantar Nodular Necrotizing Eosinophilic Fibromatosis / contracture Dupuytren's contracture Plantar fibromatosis Aggressive fibromatosis Knuckle pads
Hereditary angiopathy with nephropathy, aneurysms, and muscle cramps (HANAC) syndrome is part of a group of conditions called the COL4A1 -related disorders. ... In people with HANAC syndrome, angiopathy affects several parts of the body. ... Muscle cramps experienced by most people with HANAC syndrome typically begin in early childhood. ... Muscle cramps can be spontaneous or triggered by exercise. Individuals with HANAC syndrome also experience a variety of eye problems. ... The COL4A1 gene mutations that cause HANAC syndrome result in the production of a protein that disrupts the structure of type IV collagen.
Epidemiology The prevalence of HANAC syndrome (hereditary angiopathy-nephropathy-aneurysms-muscle cramps syndrome) is not available, but at least six affected families have been reported worldwide to date. Clinical description Typical clinical manifestations of HANAC syndrome are the presence of bilateral retinal arteriolar tortuosity (systematically observed), small-vessel brain disease (including porencephaly, leukoencephalopathy, dilated perivascular spaces, lacunar infarcts, and microbleeds, with variable clinical expression), single or multiple intracranial aneurysms (mainly located on the carotid siphon), bilateral cortical and medullary renal cysts, muscle cramps, hematuria, and persistent elevation of serum creatine kinase (CK) concentration. Other eye defects (non-syndromic autosomal dominant congenital cataract, eye anterior segment anomaly of Axenfeld-Rieger type) and glomerular filtration rate decrease have also been reported. ... Genetic counseling The pattern of inheritance of HANAC syndrome is autosomal dominant. Genetic counseling can inform parents that the risk of transmitting the mutation responsible for the disease to their children is 50%. The proportion of HANAC syndrome cases caused by a de novo pathogenic variant has been estimated to be at least 27%.
Hereditary angiopathy with nephropathy, aneurysms, and muscle cramps (HANAC) syndrome is a genetic condition that causes blood vessels to become fragile. ... While muscle cramps may begin in childhood, many of the other symptoms do not appear until later in life. HANAC syndrome is caused by mutations in the COL4A1 gene.
Gekeler et al. (2006) noted that the high degree of tortuosity of capillaries observed on nailfold capillaroscopy indicated systemic vascular pathology, but stated that the syndrome in these patients was distinct from that described by Plaisier et al. (2005). ... Plaisier et al. (2007) proposed the acronym HANAC for this syndrome. The clinical renal manifestations of the HANAC syndrome in these families included hematuria and bilateral large cysts. ... The systemic angiopathy of the HANAC syndrome appeared to affect both small vessels and large arteries. ... A suggestion by Plaisier et al. (2007) that the phenotype of the HANAC syndrome may be caused by dominant-negative effects of the mutations was supported by findings from several animal models (Gould et al., 2005; Gould et al., 2007).
Single transverse palmar crease Other names Simian crease, simian line Single transverse palmar crease on an infant's hand Specialty Medical genetics In humans, a single transverse palmar crease is a single crease that extends across the palm of the hand , formed by the fusion of the two palmar creases (known in palmistry as the "heart line" and the "head line"). It is often found in Down Syndrome, [1] but is not necessarily an indication that a person with single transverse palmar crease has the condition. ... In its non-symptomatic form, it is more common among Asians and Native Americans than among other populations, and in some families there is a tendency to inherit the condition unilaterally; that is, on one hand only. [ citation needed ] The presence of a single transverse palmar crease can be, but is not always, a symptom associated with abnormal medical conditions, such as fetal alcohol syndrome , or with genetic chromosomal abnormalities, including Down syndrome ( chromosome 21 ), cri du chat syndrome ( chromosome 5 ), Klinefelter syndrome , Wolf-Hirschhorn Syndrome , Noonan syndrome ( chromosome 12 ), Patau syndrome ( chromosome 13 ), IDIC 15/Dup15q ( chromosome 15 ), Edward's syndrome ( chromosome 18 ), and Aarskog-Scott syndrome (X-linked recessive), or autosomal recessive disorder, such as Leukocyte adhesion deficiency-2 (LAD2) . [4] A unilateral single palmar crease was also reported in a case of chromosome 9 mutation causing Nevoid basal cell carcinoma syndrome and Robinow syndrome . [5] It is also sometimes found on the hand of the affected side of patients with Poland syndrome , and craniosynostosis . ... Preventive management of children with congenital anomalies and syndromes . Cambridge, UK: Cambridge University Press. p. 147 . ... "Leukocyte adhesion deficiency II syndrome, a generalized defect in fucose metabolism" . ... "Interstitial deletion of chromosome 9, int del(9)(9q22.31-q31.2), including the genes causing multiple basal cell nevus syndrome and Robinow/brachydactyly 1 syndrome".
Hartsfield syndrome is a rare condition characterized by holoprosencephaly, which is an abnormality of brain development, and a malformation of the hands and feet called ectrodactyly. ... The FGFR1 gene mutations that cause Hartsfield syndrome severely disrupt the function of the FGFR1 protein, including its ability to bind to FGFs. ... It is unclear how these changes lead specifically to holoprosencephaly, ectrodactyly, and the other features of Hartsfield syndrome. Some people with Hartsfield syndrome do not have an identified mutation in the FGFR1 gene. ... Learn more about the gene associated with Hartsfield syndrome FGFR1 Inheritance Pattern Hartsfield syndrome can have either an autosomal dominant or autosomal recessive pattern of inheritance. ... However, in a small number of cases, people with Hartsfield syndrome have inherited the altered gene from an unaffected parent who has an FGFR1 gene mutation only in the sperm or egg cells.
A rare, genetic, multiple congenital anomalies syndrome characterized by variable expression of the holoprosenphaly (HPE) spectrum in association with ectrodactyly, cleft lip/palate and/or other ectodermal anomalies. ... There has been anecdotal reports of skull defects, vertebral anomalies, radial aplasia, eye anomalies, and cardiac malformation. Etiology Hartsfield syndrome is most often caused by the presence of heterozygous pathogenic variants in the FGFR1 gene (8p11.23), although bi-allelic pathogenic variants have been described in a minority of patients. ... Differential diagnosis The association of HPE and ectrodactyly is quite unique to Hartsfield syndrome. Phenotypic overlap can be observed in Kallman syndrome, isolated congenital hypogonadotropic hypogonadism, EEC (ectrodactyly ectodermal dysplasia and cleft lip/palate syndrome), HPE, and septo-optic dysplasia spectrum. ... Genetic counseling Depending on the variants identified, transmission of Hartsfield syndrome can follow an autosomal dominant or recessive mode of inheritance.
Summary Clinical characteristics. Hartsfield syndrome comprises two core features: holoprosencephaly (HPE) spectrum disorders and ectrodactyly spectrum disorders. ... Other brain malformations observed in persons with Hartsfield syndrome include corpus callosum agenesis, absent septum pellucidum, absent olfactory bulbs and tracts, and vermian hypoplasia. ... Diagnosis Suggestive Findings Hartsfield syndrome should be suspected in individuals with the following core features: Holoprosencephaly (HPE) spectrum disorders. ... Phenotype of Autosomal Recessive Hartsfield Syndrome Compared with four individuals with a heterozygous FGFR1 pathogenic variant, the two with biallelic FGFR1 pathogenic variants had a more severe phenotype [Simonis et al 2013]: HPE spectrum: Alobar holoprosencephaly (1/2) Diminished cortical thickening (2/2) Absent corpus callosum (2/2) Median cleft (1/2) Hypotelorism (2/2) Severe developmental delay and growth retardation (2/2) Ectrodactyly spectrum: split hand/foot malformation of both hands and feet, and fewer than three digits bilaterally (2/2) Death before age five years (2/2) Nomenclature In the older literature, Harstfield syndrome has been referred to as: Holoprosencephaly and split hand/foot syndrome Holoprosencephaly, hypertelorism, and ectrodactyly syndrome (HHES) Genotype-Phenotype Correlations No genotype-phenotype correlations have been established among the small number of affected individuals with a molecularly confirmed diagnosis of Hartsfield syndrome reported to date. ... One individual with duplication of Xq24 and Hartsfield syndrome phenotype has been reported [Takenouchi et al 2012].
A number sign (#) is used with this entry because of evidence that Hartsfield syndrome (HRTFDS) is caused by heterozygous mutation in the FGFR1 gene (136350) on chromosome 8p11. Description Hartsfield syndrome classically refers to the triad of holoprosencephaly, ectrodactyly, and cleft/lip palate. ... See also ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome (EEC; 129900), which shows phenotypic similarities. ... They noted phenotypic overlap with the EEC syndrome (see 129900). Vilain et al. (2009) reported 5 unrelated males with a phenotype most consistent with a diagnosis of Hartsfield syndrome. ... Simonis et al. (2013) noted that the fetus exhibited features that deviated substantially from those of FGFR1 mutation-positive patients with Hartsfield syndrome and might represent another diagnostic entity.
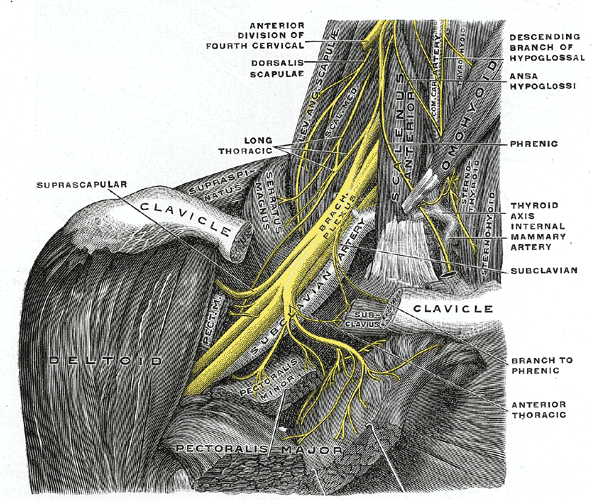
Parsonage–Turner syndrome Other names acute brachial radiculitis, [1] Parsonage–Aldren–Turner syndrome', neuralgic amyotrophy, [2] brachial neuritis, brachial plexus neuropathy, [3] brachial plexitis, acute brachial neuropathy The right brachial plexus with its short branches, viewed from in front. Specialty Neurology Parsonage–Turner syndrome , also known as acute brachial neuropathy and neuralgic amyotrophy , is a syndrome of unknown cause; although many specific risk factors have been identified (such as; post-operatively, post-infectious, post-traumatic or post-vaccination), [4] the cause is still unknown. ... Retrieved 4 November 2010 . ^ a b Frank Gaillard MD. "Parsonage-Turner syndrome" . Radiopaedia. ^ synd/1910 at Who Named It? ... "Neuralgic amyotrophy; the shoulder-girdle syndrome". Lancet . 1 (6513): 973–8. doi : 10.1016/S0140-6736(48)90611-4 . ... External links [ edit ] Classification D ICD - 10 : G54.5 ICD - 9-CM : 353.5 MeSH : D020968 DiseasesDB : 32166 v t e Diseases relating to the peripheral nervous system Mononeuropathy Arm median nerve Carpal tunnel syndrome Ape hand deformity ulnar nerve Ulnar nerve entrapment Froment's sign Ulnar tunnel syndrome Ulnar claw radial nerve Radial neuropathy Wrist drop Cheiralgia paresthetica long thoracic nerve Winged scapula Backpack palsy Leg lateral cutaneous nerve of thigh Meralgia paraesthetica tibial nerve Tarsal tunnel syndrome plantar nerve Morton's neuroma superior gluteal nerve Trendelenburg's sign sciatic nerve Piriformis syndrome Cranial nerves See Template:Cranial nerve disease Polyneuropathy and Polyradiculoneuropathy HMSN Charcot–Marie–Tooth disease Dejerine–Sottas disease Refsum's disease Hereditary spastic paraplegia Hereditary neuropathy with liability to pressure palsy Familial amyloid neuropathy Autoimmune and demyelinating disease Guillain–Barré syndrome Chronic inflammatory demyelinating polyneuropathy Radiculopathy and plexopathy Brachial plexus injury Thoracic outlet syndrome Phantom limb Other Alcoholic polyneuropathy Other General Complex regional pain syndrome Mononeuritis multiplex Peripheral neuropathy Neuralgia Nerve compression syndrome