-
Coprolalia
Wikipedia
Semantic features and Tourette's Syndrome . Retrieved 21 November 2006 . ... PMID 11794551 . ^ Singer C (May 1997). "Tourette syndrome. Coprolalia and other coprophenomena". ... PMID 1498584 . ^ Jinnah HA. Lesch-Nyhan Syndrome. eMedicine.com (August 29, 2006). Accessed 28 October 2006. ^ Tourette Syndrome FAQ. Tourette Syndrome Association. ... PMID 10972415 . ^ Singer C (May 1997). "Tourette syndrome. Coprolalia and other coprophenomena".
-
Cutis Laxa
Wikipedia
Cutis laxa is associated with deficient or absent elastin fibers in the extracellular matrix . [6] This can be related to decreased elastin synthesis or structural defects in the extracellular matrix . [7] Cutis laxa may be caused by mutations in the genes: ELN , [8] ATP6V0A2 , [9] ATP7A , [10] FBLN4 , [11] FBLN5 , [12] and PYCR1 . [13] A related neurocutaneous syndrome may be caused by mutations in the gene ALDH18A1 ( P5CS ). [14] Cutis laxa may also be seen in association with inherited connective tissue disorders such as Ehlers–Danlos syndromes . Another syndrome associated with cutis laxa is Lenz-Majewski syndrome which is due to a mutation in the phosphatidylserine synthase 1 ( PTDSS1 ) gene. ... No pharmacological agent has been able to stop the progression of the disease. [15] However, cosmetic surgeries are potentially an option as cutis laxa does not generally involve vascular fragility. [15] See also [ edit ] Occipital horn syndrome List of cutaneous conditions References [ edit ] ^ Rapini RP, Bolognia JL, Jorizzo JL (2007). ... In GeneReviews External links [ edit ] Classification D ICD - 10 : L57.4 , Q82.8 ( ILDS Q82.816) ICD - 9-CM : 701.8 , 756.83 OMIM : 123700 219100 219200 304150 MeSH : D003483 DiseasesDB : 29439 External resources eMedicine : derm/03 GeneReviews : FBLN5-Related Cutis Laxa Orphanet : 209 Medscape entry on Cutis Laxa v t e Radiation-related disorders / Photodermatoses Ultraviolet / ionizing Sunburn Phytophotodermatitis Solar urticaria Polymorphous light eruption Benign summer light eruption Juvenile spring eruption Acne aestivalis Hydroa vacciniforme Solar erythema Non-ionizing Actinic rays Actinic keratosis Atrophic actinic keratosis Hyperkeratotic actinic keratosis Lichenoid actinic keratosis Pigmented actinic keratosis Actinic cheilitis Actinic granuloma Actinic prurigo Chronic actinic dermatitis Infrared / heat Erythema ab igne ( Kangri ulcer Kairo cancer Kang cancer Peat fire cancer ) Cutis rhomboidalis nuchae Poikiloderma of Civatte Other Radiation dermatitis Acute Chronic radiodermatitis ) Favre–Racouchot syndrome Photoaging Photosensitivity with HIV infection Phototoxic tar dermatitis v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation BirthmarkFBLN5, ELN, PYCR1, EFEMP2, LTBP4, ATP7A, SLURP1, ATP6V0A2, ALDH18A1, GSN, PTDSS1, ATP6AP1, B3GALT6, GORAB, MLXIPL, B3GAT3, RPS6KA3, B4GALT7, NBAS, NAA10, SLC7A7, OSMR, CHST3, MAN1B1, CD96, IFT43, ABCC6, FAM120AOS, WDR35, GGCX, WDR19, FBN1, EZH2, COL5A1, BCL11B, MEGF8, SRD5A3, SLC2A10, LOX, MMP2, MMP9, LOXL1, SERPINA1, MBD5, ATP6V0A4, POSTN, MMRN1, AMD1, RBM19, MKKS, ATP6V1E1, CAT, COX8A, DCN, ECHS1, EGF, EPHA3, FOS, HRAS, ANOS1, MMP1, MMP3, MMP12, MMP14, PLCG2, PMM2, AMD1P2, SELPLG, KMT2D, ATP6V0D2

-
X-Linked Recessive Inheritance
Wikipedia
A minority of Alport syndrome cases are due to an autosomal recessive mutation in the gene coding for type IV collagen . Androgen insensitivity syndrome ; variable degrees of undervirilization and/or infertility in XY persons of either sex Barth syndrome ; metabolism distortion, delayed motor skills, stamina deficiency, hypotonia, chronic fatigue, delayed growth, cardiomyopathy, and compromised immune system. ... Hypohidrotic ectodermal dysplasia , presenting with hypohidrosis, hypotrichosis, hypodontia Kabuki syndrome (the KDM6A variant); multiple congenital anomalies and mental retardation. Spinal and bulbar muscular atrophy ; muscle cramps and progressive weakness Lesch–Nyhan syndrome ; neurologic dysfunction, cognitive and behavioral disturbances including self-mutilation, and uric acid overproduction (hyperuricemia) Lowe syndrome ; hydrophthalmia, cataracts, intellectual disabilities, aminoaciduria, reduced renal ammonia production and vitamin D-resistant rickets Menkes disease ; sparse and coarse hair, growth failure, and deterioration of the nervous system Nasodigitoacoustic syndrome ; misshaped nose, brachydactyly of the distal phalanges , sensorineural deafness Nonsyndromic deafness ; hearing loss Norrie disease ; cataracts, leukocoria along with other developmental issues in the eye Occipital horn syndrome ; deformations in the skeleton Ocular albinism ; lack of pigmentation in the eye Ornithine transcarbamylase deficiency ; developmental delay and mental retardation. ... External links [ edit ] X-linked diseases from the Wellcome Trust v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasia [Female X-linked disorders] https://www.nlm.nih.gov/medlineplus/ency/article/002051.htmABCB7, AMMECR1, OFD1, IKBKG, CASK, FGF16, AP1S2, AIFM1, LRAT, FRMPD4, MED12, CUL4B, MAMLD1, PQBP1, BCAP31, ATP6AP2, EBP, GJB6, MAGED2, MID2, OGT, LAGE3, ABCA4, TSPAN7, TRAPPC2, SLC6A8, SMS, SSR4, SYN1, SYP, TAF1, TAZ, UBA1, NAA10, UBE2A, CLRN1, WAS, ZIC3, USP9X, RBM10, KDM5C, SMC1A, IL1RAPL1, IQSEC2, PHF8, RAB39B, ARL6, PHF6, SLC9A7, TSR2, HS6ST2, FRMD7, CHRDL1, GPRASP2, PIH1D3, ARHGEF9, PTCHD1, C8orf37, ARX, VMA21, BRWD3, ATP11C, NEXMIF, USP27X, LAS1L, KLHL15, POF1B, ALG13, AIPL1, KIF4A, FTSJ1, RBMX, CCDC22, SH3KBP1, NSDHL, FOXP3, RLIM, MBTPS2, TMLHE, ZC4H2, TEX11, THOC2, NYX, BCORL1, UPF3B, RS1, RPL10, RPGR, EIF2S3, CRX, CYBB, DDX3X, TIMM8A, DKC1, DLG3, DMD, EDA, EMD, GATA1, F8, F9, FANCB, GPC4, FGD1, FHL1, FLNA, AFF2, COL4A6, CNGA1, CLIC2, CLCN5, NR0B1, ALAS2, ABCD1, XIAP, AR, STS, ARSL, ATP2B3, ATP6AP1, ATP7A, ATRX, AVPR2, BGN, BTK, CACNA1F, CD40LG, CLCN4, G6PD, OPN1MW, ROM1, PGK1, NDUFA1, NONO, OCRL, OPHN1, OTC, PAK3, PDE6G, CFP, PHKA1, GJB1, PHKA2, PIGA, PLP1, POLA1, POU3F4, PRPS1, RBP3, OPN1LW, NDP, MTM1, MSN, MID1, GJB2, GK, GLA, GPC3, GRIA3, HCFC1, HPRT1, IDS, IGBP1, IGF2, IGSF1, IL2RG, ANOS1, L1CAM, SH2D1A, MAOA, MECP2, GPR179

-
Chromosomal Deletion Syndrome
Wikipedia
Chromosomal deletion syndrome An example of chromosomal deletions Chromosomal deletion syndromes result from deletion of parts of chromosomes . ... Smaller deletions result in Microdeletion syndrome , which are detected using fluorescence in situ hybridization (FISH) Examples of chromosomal deletion syndromes include 5p -Deletion ( cri du chat syndrome ), 4p -Deletion ( Wolf-Hirschhorn syndrome ), Prader–Willi syndrome , and Angelman syndrome . [1] Contents 1 5p-Deletion 2 4p-Deletion 3 Prader-Willi vs. Angelman Syndrome 4 See also 5 References 5p-Deletion [ edit ] The chromosomal basis of Cri du chat syndrome consists of a deletion of the most terminal portion of the short arm of chromosome 5. 5p deletions, whether terminal or interstitial, occur at different breakpoints; the chromosomal basis generally consists of a deletion on the short arm of chromosome 5. ... "Comparative molecular approaches in Prader-Willi syndrome diagnosis". Gene . 575 (2 Pt 1): 353–8. doi : 10.1016/j.gene.2015.08.058 . ... PMID 26335514 . ^ Cassidy, Suzanne B.; Schwartz, Stuart; Miller, Jennifer L.; Driscoll, Daniel J. (2012-01-01). "Prader-Willi syndrome" . Genetics in Medicine . 14 (1): 10–26. doi : 10.1038/gim.0b013e31822bead0 .

-
Rabbit Syndrome
Wikipedia
Rabbit syndrome is a rare [1] form of extrapyramidal side effect of antipsychotic drugs in which perioral tremors occur at a rate of approximately 5 Hz. ... PMID 2870650 . ^ Villeneuve, A (1972). "The rabbit syndrome. A peculiar extrapyramidal reaction" . ... "Newer antipsychotics and the rabbit syndrome" . Clinical Practice and Epidemiology in Mental Health . 3 : 6. doi : 10.1186/1745-0179-3-6 . ... PMID 17562001 . ^ Schwartz, M; Hocherman, S (2004). "Antipsychotic-induced rabbit syndrome: Epidemiology, management and pathophysiology". ... S2CID 11451531 . ^ Gonidakis, F; Ploubidis, D; Papadimitriou, G (2008). "Aripiprazole-induced rabbit syndrome in a drug-naive schizophrenic patient".
-
Asperger Syndrome, X-Linked, Susceptibility To, 2
Omim
A number sign (#) is used with this entry because of evidence that X-linked Asperger syndrome-2 (ASPGX2) is conferred by variation mutation in the NLGN4 gene (300427) on chromosome Xp22. Description Asperger syndrome is considered to be a form of childhood autism (see, e.g., 209850). The DSM-IV (American Psychiatric Association, 1994) specifies several diagnostic criteria for Asperger syndrome, which has many of the same features as autism. ... Gillberg et al. (2001) described the development of the Asperger syndrome (and high-functioning autism) Diagnostic Interview (ASDI), which they claimed has a strong validity in the diagnosis of the disorder. For a discussion of genetic heterogeneity of Asperger syndrome, see 608638. Molecular Genetics In a boy with X-linked Asperger syndrome, Jamain et al. (2003) identified a mutation in the NLGN4 gene (300427.0001).
-
Carney-Stratakis Syndrome
Orphanet
Carney-Stratakis syndrome is a recently described familial syndrome characterized by gastrointestinal stromal tumors (GIST) and paragangliomas, often at multiple sites. Epidemiology It is a very rare syndrome reported in less than 20 unrelated families to date. ... Clinical description Patients with Carney-Stratakis syndrome have both GIST and paraganglioma. ... Targeting SDH function may potentiallybe useful in treating Carney-Stratakis syndrome patients but, at present, there are no drugs that restore SDH function. Life-long follow-up should be offered to patients with Carney-Stratakis syndrome.
-
Char Syndrome
Gene_reviews
Diagnosis Formal clinical diagnostic criteria for Char syndrome have not been published. Suggestive Findings Char syndrome should be suspected in individuals with the following clinical and family history findings. ... Penetrance The penetrance of Char syndrome has not been formally determined. ... Prevalence The prevalence of Char syndrome has not been determined but is thought to be quite low. ... The hand anomalies associated with Char syndrome can be as minimal as fifth finger clinodactyly, which can be a normal finding and overlaps with numerous other syndromes. ... Tabatznik syndrome [Silengo et al 1990] Heart-hand syndrome type III (OMIM 140450) Management Evaluations Following Initial Diagnosis To establish the extent of disease and needs in an individual diagnosed with Char syndrome, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
-
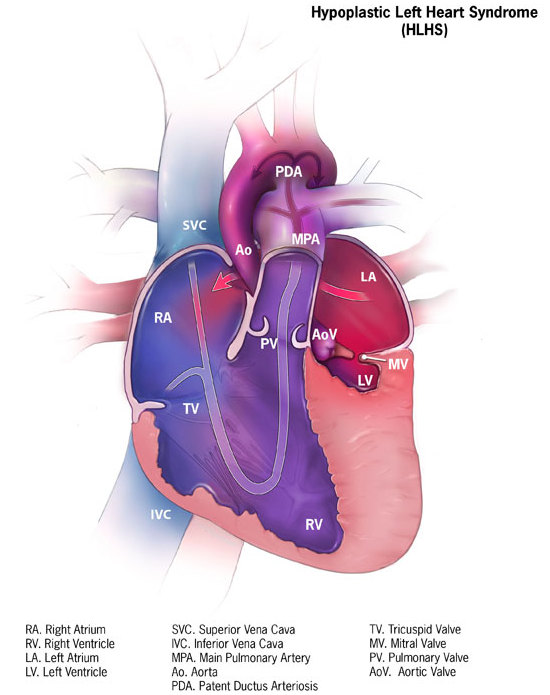
Hypoplastic Left Heart Syndrome
Wikipedia
Hypoplastic left heart syndrome Other names Cyanotic heart disease - hypoplastic left heart [1] Illustration of heart suffering from hypoplastic left heart syndrome Specialty Cardiology Hypoplastic left heart syndrome ( HLHS ) is a rare congenital heart defect in which the left side of the heart is severely underdeveloped. ... "Seasonality of hypoplastic left heart syndrome in the United States: a 10-year time-series analysis". ... "Hypoplastic left heart syndrome: current considerations and expectations" . ... "Hybrid approach for hypoplastic left heart syndrome: intermediate results after the learning curve". ... CS1 maint: multiple names: authors list ( link ) External links [ edit ] Hypoplastic left heart syndrome information for parents. Classification D ICD - 10 : Q23.4 ICD - 9-CM : 746.7 OMIM : 241550 MeSH : D018636 DiseasesDB : 31507 External resources MedlinePlus : 001106 eMedicine : ped/1131 Orphanet : 2248 Wikimedia Commons has media related to Hypoplastic left heart syndrome . v t e Congenital heart defects Heart septal defect Aortopulmonary septal defect Double outlet right ventricle Taussig–Bing syndrome Transposition of the great vessels dextro levo Persistent truncus arteriosus Aortopulmonary window Atrial septal defect Sinus venosus atrial septal defect Lutembacher's syndrome Ventricular septal defect Tetralogy of Fallot Atrioventricular septal defect Ostium primum Consequences Cardiac shunt Cyanotic heart disease Eisenmenger syndrome Valvular heart disease Right pulmonary valves stenosis insufficiency absence tricuspid valves stenosis atresia Ebstein's anomaly Left aortic valves stenosis insufficiency bicuspid mitral valves stenosis regurgitation Other Underdeveloped heart chambers right left Uhl anomaly Dextrocardia Levocardia Cor triatriatum Crisscross heart Brugada syndrome Coronary artery anomaly Anomalous aortic origin of a coronary artery Ventricular inversion v t e Diseases of ion channels Calcium channel Voltage-gated CACNA1A Familial hemiplegic migraine 1 Episodic ataxia 2 Spinocerebellar ataxia type-6 CACNA1C Timothy syndrome Brugada syndrome 3 Long QT syndrome 8 CACNA1F Ocular albinism 2 CSNB2A CACNA1S Hypokalemic periodic paralysis 1 Thyrotoxic periodic paralysis 1 CACNB2 Brugada syndrome 4 Ligand gated RYR1 Malignant hyperthermia Central core disease RYR2 CPVT1 ARVD2 Sodium channel Voltage-gated SCN1A Familial hemiplegic migraine 3 GEFS+ 2 Febrile seizure 3A SCN1B Brugada syndrome 6 GEFS+ 1 SCN4A Hypokalemic periodic paralysis 2 Hyperkalemic periodic paralysis Paramyotonia congenita Potassium-aggravated myotonia SCN4B Long QT syndrome 10 SCN5A Brugada syndrome 1 Long QT syndrome 3 SCN9A Erythromelalgia Febrile seizure 3B Paroxysmal extreme pain disorder Congenital insensitivity to pain Constitutively active SCNN1B / SCNN1G Liddle's syndrome SCNN1A / SCNN1B / SCNN1G Pseudohypoaldosteronism 1AR Potassium channel Voltage-gated KCNA1 Episodic ataxia 1 KCNA5 Familial atrial fibrillation 7 KCNC3 Spinocerebellar ataxia type-13 KCNE1 Jervell and Lange-Nielsen syndrome Long QT syndrome 5 KCNE2 Long QT syndrome 6 KCNE3 Brugada syndrome 5 KCNH2 Short QT syndrome KCNQ1 Jervell and Lange-Nielsen syndrome Romano–Ward syndrome Short QT syndrome Long QT syndrome 1 Familial atrial fibrillation 3 KCNQ2 BFNS1 Inward-rectifier KCNJ1 Bartter syndrome 2 KCNJ2 Andersen–Tawil syndrome Long QT syndrome 7 Short QT syndrome KCNJ11 TNDM3 KCNJ18 Thyrotoxic periodic paralysis 2 Chloride channel CFTR Cystic fibrosis Congenital absence of the vas deferens CLCN1 Thomsen disease Myotonia congenita CLCN5 Dent's disease CLCN7 Osteopetrosis A2, B4 BEST1 Vitelliform macular dystrophy CLCNKB Bartter syndrome 3 TRP channel TRPC6 FSGS2 TRPML1 Mucolipidosis type IV Connexin GJA1 Oculodentodigital dysplasia Hallermann–Streiff syndrome Hypoplastic left heart syndrome GJB1 Charcot–Marie–Tooth disease X1 GJB2 Keratitis–ichthyosis–deafness syndrome Ichthyosis hystrix Bart–Pumphrey syndrome Vohwinkel syndrome ) GJB3 / GJB4 Erythrokeratodermia variabilis Progressive symmetric erythrokeratodermia GJB6 Clouston's hidrotic ectodermal dysplasia Porin AQP2 Nephrogenic diabetes insipidus 2 See also: ion channelsNKX2-5, GJA1, SAP130, PCDHA13, PCDHA9, NOTCH1, FOXF1, MYRF, ARHGAP31, TBX1, TBX5, NR2F2, WT1, ZNF148, TBX20, CRELD1, ARID1A, KYNU, WASHC5, HAAO, MKKS, PAH, CCDC22, SMAD6, GATA6, GATA4, FLI1, TRAF7, GATA5, B3GLCT, DTNA, PKD1L1, TMEM258, MCTP2, HAND1, MYH6, JAM3, C20orf181, APOE, FOXP1, RBFOX2, KMT2D, SMN2, S100B, PROX1, FXN, FOXC2, FOXL1, EDN1, ACE, CBL, RN7SL263P
-
Miller-Fisher Syndrome
Gard
Miller Fisher syndrome is a rare acquired nerve disease considered to be a variant of Guillain-Barré syndrome . ... In most people with Miller Fisher syndrome an antibody ( anti-GQ1b ) is identified. The presence of these autoantibodies helps confirm the diagnosis of the syndrome. Treatment includes intravenous immunoglobulin (IVIG), plasmapheresis (a plasma exchange procedure in which the antibodies are removed from the blood) and supportive care. The prognosis is usually good, and in most cases, there is almost complete recovery within 6 months. In rare cases, the syndrome may progress and permanent neurological deficits may be present.
-
Shaken Baby Syndrome
Mayo_clinic
Overview Shaken baby syndrome is a serious brain injury resulting from forcefully shaking an infant or toddler. It's also known as abusive head trauma, shaken impact syndrome, inflicted head injury or whiplash shaken infant syndrome. Shaken baby syndrome destroys a child's brain cells and prevents his or her brain from getting enough oxygen. ... In mild cases of shaken baby syndrome, a child may appear normal after being shaken, but over time they may develop health or behavioral problems. ... Shaken baby syndrome isn't usually caused by bouncing a child on your knee or minor falls.

-
Wellens' Syndrome
Wikipedia
Wellens' syndrome Other names Wellens' sign, Wellens' warning, Wellens' waves EKG of a 69-year-old black male with Wellens' syndrome. ... A subsequent prospective study identified this syndrome in 14% of patients at presentation and 60% of patients within the first 24 hours. [3] The presence of Wellens' syndrome carries significant diagnostic and prognostic value. ... In the original Wellens' study group, 75% of those with the typical syndrome manifestations had an anterior myocardial infarction. ... EKG/ECG in someone with Wellens' syndrome when having chest pain EKG/ECG of the same person when pain-free, note the biphasic T waves in leads V2 and V3 References [ edit ] ^ Tandy, TK; Bottomy DP; Lewis JG (March 1999). "Wellens' syndrome" . Annals of Emergency Medicine . 33 (3): 347–351. doi : 10.1016/S0196-0644(99)70373-2 .

-
Nevus Anemicus
Wikipedia
J Am Acad Dermatol 1997;37:523-49, quiz 549-52. v t e Pigmentation disorders / Dyschromia Hypo- / leucism Loss of melanocytes Vitiligo Quadrichrome vitiligo Vitiligo ponctué Syndromic Alezzandrini syndrome Vogt–Koyanagi–Harada syndrome Melanocyte development Piebaldism Waardenburg syndrome Tietz syndrome Loss of melanin / amelanism Albinism Oculocutaneous albinism Ocular albinism Melanosome transfer Hermansky–Pudlak syndrome Chédiak–Higashi syndrome Griscelli syndrome Elejalde syndrome Griscelli syndrome type 2 Griscelli syndrome type 3 Other Cross syndrome ABCD syndrome Albinism–deafness syndrome Idiopathic guttate hypomelanosis Phylloid hypomelanosis Progressive macular hypomelanosis Leukoderma w/o hypomelanosis Vasospastic macule Woronoff's ring Nevus anemicus Ungrouped Nevus depigmentosus Postinflammatory hypopigmentation Pityriasis alba Vagabond's leukomelanoderma Yemenite deaf-blind hypopigmentation syndrome Wende–Bauckus syndrome Hyper- Melanin / Melanosis / Melanism Reticulated Dermatopathia pigmentosa reticularis Pigmentatio reticularis faciei et colli Reticulate acropigmentation of Kitamura Reticular pigmented anomaly of the flexures Naegeli–Franceschetti–Jadassohn syndrome Dyskeratosis congenita X-linked reticulate pigmentary disorder Galli–Galli disease Revesz syndrome Diffuse/ circumscribed Lentigo / Lentiginosis : Lentigo simplex Liver spot Centrofacial lentiginosis Generalized lentiginosis Inherited patterned lentiginosis in black persons Ink spot lentigo Lentigo maligna Mucosal lentigines Partial unilateral lentiginosis PUVA lentigines Melasma Erythema dyschromicum perstans Lichen planus pigmentosus Café au lait spot Poikiloderma ( Poikiloderma of Civatte Poikiloderma vasculare atrophicans ) Riehl melanosis Linear Incontinentia pigmenti Scratch dermatitis Shiitake mushroom dermatitis Other/ ungrouped Acanthosis nigricans Freckle Familial progressive hyperpigmentation Pallister–Killian syndrome Periorbital hyperpigmentation Photoleukomelanodermatitis of Kobori Postinflammatory hyperpigmentation Transient neonatal pustular melanosis Other pigments Iron Hemochromatosis Iron metallic discoloration Pigmented purpuric dermatosis Schamberg disease Majocchi's disease Gougerot–Blum syndrome Doucas and Kapetanakis pigmented purpura / Eczematid-like purpura of Doucas and Kapetanakis Lichen aureus Angioma serpiginosum Hemosiderin hyperpigmentation Other metals Argyria Chrysiasis Arsenic poisoning Lead poisoning Titanium metallic discoloration Other Carotenosis Tar melanosis Dyschromia Dyschromatosis symmetrica hereditaria Dyschromatosis universalis hereditaria See also Skin color Skin whitening Tanning Sunless Tattoo removal Depigmentation

-
Neu–laxova Syndrome
Wikipedia
Unsourced material may be challenged and removed. Find sources: "Neu–Laxova syndrome" – news · newspapers · books · scholar · JSTOR ( December 2018 ) ( Learn how and when to remove this template message ) Neu–Laxova syndrome Other names Neu-Povysilová syndrome Specialty Medical genetics Neu–Laxova syndrome (also known as Neu syndrome or Neu-Povysilová syndrome , abbreviated as NLS) is a rare autosomal recessive disorder characterized by severe intrauterine growth restriction and multiple congenital malformations . Neu–Laxova syndrome is a very severe disorder, leading to stillbirth or death shortly after birth. ... Renata Laxova in 1972 [2] as a lethal disorder in siblings with multiple malformations. Neu–Laxova syndrome is an extremely rare disorder with less than 100 cases reported in medical literature. ... The longest survival reported in literature is of 134 days. This syndrome is transmitted as an autosomal recessive disorder and there is a risk for recurrence of 25% in future pregnancies. References [ edit ] ^ Neu, Richard L.; Kajii, Tadashi; Gardner, Lytt I.; Nagyfy, Stephen F.; King, Saddie (March 1, 1971). "A Lethal Syndrome of Microcephaly with Multiple Congenital Anomalies in Three Siblings" .
-
5q31.3 Microdeletion Syndrome
Medlineplus
5q31.3 microdeletion syndrome is a condition characterized by severely delayed development of speech and motor skills, such as walking. ... Breathing problems and difficulty swallowing (dysphagia) can be life-threatening. 5q31.3 microdeletion syndrome is also characterized by distinctive facial features. ... Recurrent seizures (epilepsy) and seizure-like episodes (which can include muscle jerking, twitching, and stiffening), are common in 5q31.3 microdeletion syndrome. Many individuals with 5q31.3 microdeletion syndrome have brain abnormalities, several of which are caused by reduced production of myelin or delayed maturation of myelin. ... Causes 5q31.3 microdeletion syndrome is caused by a chromosomal change in which a small piece of chromosome 5 is deleted in each cell. ... It is unclear how the loss of other genes in the deleted region contributes to the development of 5q31.3 microdeletion syndrome. Learn more about the gene and chromosome associated with 5q31.3 microdeletion syndrome PURA chromosome 5 Additional Information from NCBI Gene: NRG2 Inheritance Pattern 5q31.3 microdeletion syndrome follows an autosomal dominant inheritance pattern , which means one copy of the genetic alteration in each cell is sufficient to cause the disorder.
-
Lesch-Nyhan Syndrome
Medlineplus
Lesch-Nyhan syndrome is a condition that occurs almost exclusively in males. ... People with Lesch-Nyhan syndrome usually cannot walk, require assistance sitting, and generally use a wheelchair. Self-injury (including biting and head banging) is the most common and distinctive behavioral problem in individuals with Lesch-Nyhan syndrome. Frequency The prevalence of Lesch-Nyhan syndrome is approximately 1 in 380,000 individuals. ... The signs and symptoms of Lesch-Nyhan variant are often milder than those of Lesch-Nyhan syndrome and do not include self-injury. Learn more about the gene associated with Lesch-Nyhan syndrome HPRT1 Inheritance Pattern This condition is inherited in an X-linked recessive pattern .
-
Warsaw Breakage Syndrome
Wikipedia
Warsaw breakage syndrome Other names WABS [1] Warsaw breakage syndrome is a rare genetic condition. ... Diagnosis [ edit ] The diagnosis may be suspected on clinical grounds and can be confirmed by sequencing the DDX11 gene. [ citation needed ] Differential diagnosis [ edit ] The DDX should be based on the following: [ citation needed ] Bloom syndrome Cornelia de Lange syndrome Fanconi anemia Nijmegen breakage syndrome Roberts syndrome Xeroderma pigmentosum Treatment [ edit ] There is no known curative treatment for this condition presently. Management is supportive. [ citation needed ] History [ edit ] This condition was first described in 2010. [4] References [ edit ] ^ "OMIM Entry - # 613398 - WARSAW BREAKAGE SYNDROME; WABS" . omim.org . Retrieved 29 October 2019 . ^ Alkhunaizi E, Shaheen R, Bharti SK, Joseph-George AM, Chong K, Abdel-Salam GMH, Alowain M, Blaser SI, Papsin BC, Butt M, Hashem M, Martin N, Godoy R, Brosh RM Jr, Alkuraya FS, Chitayat D (2018) Warsaw breakage syndrome: Further clinical and genetic delineation. Am J Med Genet A doi: 10.1002/ajmg.a.40482 ^ Pisani FM (2019) Spotlight on Warsaw Breakage Syndrome. Appl Clin Genet 12:239-248 ^ van der Lelij P, Chrzanowska KH, Godthelp BC, Rooimans MA, Oostra AB, Stumm M, Zdzienicka MZ, Joenje H, de Winter JP (2010) Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1.
-
Pseudo Gray Platelet Syndrome
Wikipedia
Pseudo gray platelet syndrome Specialty Hematology Pseudo gray platelet syndrome was described by Cockbill, Burmester, and Heptinstall (1988) [1] who reported a 25-year-old woman with a history of mild bruising and bleeding. ... Few cases have been reported in the literature. [ citation needed ] Diagnosis [ edit ] Comparison to gray platelet syndrome [ edit ] Pseudo-gray platelet syndrome differs from gray platelet syndrome (GPS), one of the giant platelet syndromes . [3] GPS is characterized by " thrombocytopenia , abnormally large agranular platelets in peripheral blood smears, and almost total absence of platelet alpha-granules and their constituents." [4] The defect in GPS is the failure of megakaryocytes to package secretory proteins into alpha-granules. ... You can help by adding to it . ( October 2017 ) References [ edit ] ^ Cockbill SR, Burmester HB, Heptinstall S (October 1988). "Pseudo grey platelet syndrome--grey platelets due to degranulation in blood collected into EDTA". ... PMID 3143601 . ^ Toyota S, Nakamura N, Dan K (November 2002). "Pseudo gray platelet syndrome in a patient with acute myocardial infarction". ... PMID 7957801 . ^ Jantunen E, Hänninen A, Naukkarinen A, Vornanen M, Lahtinen R (July 1994). "Gray platelet syndrome with splenomegaly and signs of extramedullary hematopoiesis: a case report with review of the literature".
-
Aase Syndrome
Wikipedia
Aase syndrome Other names Hydrocephalus-cleft palate-joint contractures syndrome, Aase-Smith syndrome Aase syndrome or Aase–Smith syndrome is a rare inherited disorder characterized by anemia with some joint and skeletal deformities. Aase syndrome is thought to be an autosomal dominant inherited disorder. [1] The genetic basis of the disease is not known. ... Cause [ edit ] Some cases of Aase syndrome (45%) have been shown to be inherited, and are due to a change in one gene which makes ribosomal proteins. ... A bone marrow transplant may be necessary if other treatment fails. [ citation needed ] Prognosis [ edit ] Anemia usually resolves over the years. [ citation needed ] References [ edit ] ^ http://www.rarediseases.org/search/rdbdetail_abstract.html?disname=Aase%20Syndrome "Aase Syndrome" ^ Aase JM, Smith DW (1968). "Dysmorphogenesis of joints, brain, and palate: a new dominantly inherited syndrome". J Pediatr . 73 (4): 606–9. doi : 10.1016/S0022-3476(68)80278-1 . PMID 5678002 . ^ "Aase syndrome: MedlinePlus Medical Encyclopedia" . medlineplus.gov .

-
Baraitser-Winter Syndrome
Medlineplus
Baraitser-Winter syndrome is a condition that affects the development of many parts of the body, particularly the face and the brain . ... Rarely, people with Baraitser-Winter syndrome have involuntary muscle tensing (dystonia). Frequency Baraitser-Winter syndrome is a rare condition. Fewer than 50 cases have been reported in the medical literature. Causes Baraitser-Winter syndrome can result from mutations in either the ACTB or ACTG1 gene. ... These changes underlie the variety of signs and symptoms associated with Baraitser-Winter syndrome. Learn more about the genes associated with Baraitser-Winter syndrome ACTB ACTG1 Inheritance Pattern This condition is described as autosomal dominant , which means one copy of the altered gene in each cell is sufficient to cause the disorder.







