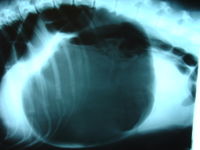
Journal of the American Animal Hospital Association . 42 (1): 28–36. doi : 10.5326/0420028 . PMID 16397192 . ^ a b "Gastric Dilatation-volvulus" . ... PMID 12941556 . ^ Bloat | DogTime - Warning signs to watch for ^ Lucas, D (15 September 2014). " ' Bloat' refers to 2 different stomach ailments in pets" . USA TODAY . Retrieved 2017-07-28 . ^ "The Simpsons - 'Dog of Death ' " . cwsanfrancisco.cbslocal.com .
CyberPsychology & Behavior . 1 : 25–28. doi : 10.1089/cpb.1998.1.25 . ^ Whang, Leo Sang-Min; Lee, Sujin; Chang, Geunyoung (2003). ... "A dynamic longitudinal examination of social media use, needs, and gratifications among college students". Computers in Human Behavior . 28 (5): 1829–1839. doi : 10.1016/j.chb.2012.05.001 . ^ Morahan-Martin, J.; Schumacher, P. (2003).
"The natural history of calf vein thrombosis: lysis of thrombi and development of reflux". Journal of Vascular Surgery . 28 (1): 67–73, discussion 73–4. doi : 10.1016/s0741-5214(98)70201-0 . ... Journal of Thrombosis and Thrombolysis . 28 (4): 465–76. doi : 10.1007/s11239-009-0309-3 .
They are about the same size as those of Heterophyes and Clonorchis , usually measuring 26-28 μm length and 15-17μm width. The egg also has a very slight opercular shoulder , marking the line of cleavage between the shell and operculum , an "escape hatch" for the mircidium . ... Lee, Soo-ung et al. "Sequence comparisons of 28S ribosomal DNA and mitochondrial cytochrome c oxidase subunit I of Metgonimus yokogawai, M. takahashii, and M. miyatai."
. ^ Tibbetts, Paul. " Symbolic Interaction Theory and the Cognitively Disabled: A neglected Dimension." Jstor. Winter 2004. Web. 28 September 2011 Symbolic Interaction Theory and the Cognitively Disabled: A Neglected Dimension ^ Mojtabai R (September 1994). "Fregoli syndrome". Aust N Z J Psychiatry . 28 (3): 458–62. doi : 10.3109/00048679409075874 .
Archived from the original (PDF) on 2008-02-27 . Retrieved 2008-01-28 . Cite journal requires |journal= ( help ) , which cites: Greenslade, Forrest; Janie Benson; Judith Winkler; Victoria Henderson; Ann Leonard (1993). ... Archived from the original on October 28, 2008 . Retrieved February 18, 2006 . ^ "All About the Machine Vacuum Aspiration Procedure for Early Abortion" . about.com .
Most of these injuries were also paid through private insurance (170: 50.63%) and Medicare (70: 20.85%). [27] The average estimated cost for the surgery also known as Tommy John surgery is $21,563. [28] References [ edit ] ^ Hoffman, James K.; Protzman, Nicole M.; Malhotra, Amit D. (2015). ... The American Journal of Sports Medicine . 28 (1): 16–23. doi : 10.1177/03635465000280011401 . ^ Longman, Jere.
The estimates range from "exceedingly rare" [18] to one in ten people with autism having savant skills in varying degrees. [1] A 2009 British study of 137 parents of autistic children found that 28% believe their children met the criteria for a savant skill, defined as a skill or power "at a level that would be unusual even for 'normal' people". [19] As many as 50 cases of sudden or acquired savant syndrome have been reported. [20] [21] Males with savant syndrome outnumber females by roughly 6:1 (in Finland), [22] slightly higher than the sex ratio disparity for autism spectrum disorders of 4.3:1. [23] History [ edit ] The term idiot savant ( French for "learned idiot") was first used to describe the condition in 1887 by John Langdon Down , who is known for his description of Down syndrome . ... "The epidemiology of autism spectrum disorders" . Annual Review of Public Health . 28 : 235–58. doi : 10.1146/annurev.publhealth.28.021406.144007 .
Bellanne-Chantelot et al. (2005) found molecular alterations of the TCF2 gene in 28 (70%) of 40 unrelated patients with a clinical phenotype consistent with MODY5. Point mutations were identified in 18 of the 28 patients and 10 had gross genomic rearrangements, which in 9 patients involved a deletion of at least 1.2 Mb, delimited by the TCF2 and TRIP3 (604500) genes and encompassing 7 other genes and 2 predicted proteins; 1 patient had a single-exon deletion of exon 5.
Renal cysts and diabetes syndrome (RCAD) is a rare form of maturity-onset diabetes of the young (MODY; see this term) characterized clinically by heterogeneous cystic renal disease and early-onset familial non-autoimmune diabetes. Pancreatic atrophy, liver dysfunction and genital tract anomalies are also features of the syndrome. Epidemiology RCAD accounts for 1-5% of MODY cases, depending on whether cases are ascertained in those with renal disease. There is an equal female to male ratio. Clinical description Patients with RCAD typically present with diabetes between 10 and 40 years of age. Unlike patients with HNF1A mutations, they are not sensitive to sulphonylureas.
A number sign (#) is used with this entry because Fanconi renotubular syndrome-4 with maturity-onset diabetes of the young (FRTS4) is caused by heterozygous mutation in the HNF4A gene (600281) on chromosome 20q13. Heterozygous mutation in the HNF4A gene can also cause isolated MODY1 (125850). For a general phenotypic description and a discussion of genetic heterogeneity of Fanconi renotubular syndrome, see FRTS1 (134600). Clinical Features Stanescu et al. (2012) reported a female infant who presented with macrosomia and severe hypoglycemia associated with hyperinsulinism in the first hours of life. Over the following years, she required lower doses to maintain normoglycemia, and treatment was discontinued at age 4 years.
Renal cysts and diabetes syndrome Other names MODY 5 MODY 5 is inherited in an autosomal dominant manner. Renal cysts and diabetes syndrome ( RCAD ), also known as MODY 5 , is a form of maturity onset diabetes of the young . HNF1β-related MODY is one of the less common forms of MODY, with some distinctive clinical features, including atrophy of the pancreas and several forms of renal disease. HNF1β, also known as transcription factor 2 (TCF2), is involved in early stages of embryonic development of several organs, including the pancreas, where it contributes to differentiation of pancreatic endocrine Ngn3 + cell progenitors from non-endocrine embryonic duct cells. The gene is on chromosome 17q . [1] The degree of insulin deficiency is variable.
These cases can be cured by removing much less of the pancreas, resulting in excellent outcomes with no long-term problems. [4] [5] [21] History [ edit ] This condition has been referred to by a variety of names in the past 50 years; nesidioblastosis and islet cell adenomatosis were favored in the 1970s, beta cell dysregulation syndrome or dysmaturation syndrome in the 1980s, and persistent hyperinsulinemic hypoglycemia of infancy (PHHI) in the 1990s. [ citation needed ] See also [ edit ] Hyperammonemia List of congenital disorders References [ edit ] ^ a b c "Familial hyperinsulinism | Genetic and Rare Diseases Information Center(GARD) – an NCATS Program" . rarediseases.info.nih.gov . Retrieved 28 November 2016 . ^ a b c Reference, Genetics Home. ... Retrieved 2017-01-27 . ^ a b c d Yorifuji, Tohru (28 November 2016). "Congenital hyperinsulinism: current status and future perspectives" .
A rare, congenital, isolated hyperinsulinism disorder characterized by neonatal presentation of severe refractory hypoglycemia in the first two days of life, with limited response to medical management, sometimes requiring pancreatic resection. Newborns are often large for gestational age with mild to moderate hepatomegaly and diffuse form of hyperinsulinism due to Kir6.2 deficiency. Persistent hypoglycemia, hyperglycemia and type1 diabetes mellitus may develop later in life. Life-threatening hypoglycemic coma or status epilepticus have also been associated.
A form of diazoxide-sensitive diffuse hyperinsulinism (DHI) characterized by hypoglycemic episodes that are usually mild, escaping detection during infancy, and usually present a good clinical response to diazoxide. Autosomal dominant hyperinsulinism due to SUR1 deficiency usually has a milder phenotype when compared to that resulting from recessive K-ATP mutations (recessive forms of Diazoxide-resistant hyperinsulinism).
Summary The purpose of this overview is to increase the awareness of clinicians regarding familial hyperinsulinism (referred to as FHI in this GeneReview ) and its genetic causes and management. The following are the goals of this overview. Goal 1. Describe the clinical characteristics of FHI. Goal 2. Review the genetic causes of FHI. Goal 3. Provide an evaluation strategy to identify the genetic cause of FHI in a proband (when possible). Goal 4. Inform (when possible) medical management of FHI based on genetic cause and evaluation of relatives at risk. Goal 5. Inform risk assessment and surveillance of at-risk relatives for early detection and treatment of FHI.
Congenital hyperinsulinism is a condition that causes individuals to have abnormally high levels of insulin, which is a hormone that helps control blood sugar levels. People with this condition have frequent episodes of low blood sugar (hypoglycemia). In infants and young children, these episodes are characterized by a lack of energy (lethargy), irritability, or difficulty feeding. Repeated episodes of low blood sugar increase the risk for serious complications such as breathing difficulties, seizures, intellectual disability, vision loss, brain damage, and coma. The severity of congenital hyperinsulinism varies widely among affected individuals, even among members of the same family.
Woolf et al. (1991) found consanguineous parentage in 5 of 28 families. Segregation analysis showed that the ratio of subsequent affected to unaffected sibs was similar to that expected of an autosomal recessive condition. ... Meissner et al. (1999) reported that in the autosomal recessive form of hyperinsulinism, 28 different mutations in the SUR1 gene and 2 in the KCNJ11 gene had been identified.
A rare, congenital, isolated hyperinsulinism disorder characterized by neonatal presentation of severe refractory hypoglycemia in the first two days of life, with limited response to medical management, sometimes requiring pancreatic resection. Newborns are often large for gestational age with mild to moderate hepatomegaly and diffuse form of hyperinsulinism due to SUR1 deficiency. Persistent hypoglycemia, hyperglycemia and type1 diabetes mellitus may develop later in life. Life-threatening hypoglycemic coma or status epilepticus have also been associated.
A rare, congenital, isolated hyperinsulinism disorder characterized by diazoxide unresponsive recurrent episodes of hyperinsulinemic hypoglycemia resulting from an excessive insulin secretion by the pancreatic bêta-cells due to Kir6.2 deficiency. Hypoglycemia may lead to variable clinical manifestation, ranging from asymptomatic hypoglycemia revealed by routine blood glucose monitoring to macrosomia at birth, mild to moderate hepatomegaly and life-threatening hypoglycemic coma or status epilepticus, further leading to poor neurological outcome.
A rare, congenital, isolated hyperinsulinism disorder characterized by diazoxide unresponsive recurrent episodes of hyperinsulinemic hypoglycemia resulting from an excessive insulin secretion by the pancreatic bêta-cells due to SUR1 deficiency. Hypoglycemia may lead to variable clinical manifestations, ranging from asymptomatic hypoglycemia revealed by routine blood glucose monitoring to macrosomia at birth, mild to moderate hepatomegaly and life-threatening hypoglycemic coma or status epilepticus, further leading to poor neurological outcome.
A form of diazoxide-sensitive diffuse hyperinsulinism (DHI) characterized by hypoglycemic epiosodes that are usually mild, escaping detection during infancy, and usually a good clinical response to diazoxide, (but some are diazoxide resistant). Autosomal dominant hyperinsulinism due to Kir6.2 deficiency usually has a milder phenotype when compared to that resulting from recessive K+ (K-ATP) channel mutations (Recessive forms of diazoxide-resistant hyperinsulinism).
A number sign (#) is used with this entry because familial hyperinsulinemic hypoglycemia-2 is caused by mutation in the gene encoding the Kir6.2 subunit of the inwardly rectifying potassium channel (KCNJ11; 600937). For a phenotypic description and a discussion of genetic heterogeneity of hyperinsulinemic hypoglycemia, see HHF1 (256450). Clinical Features Nestorowicz et al. (1997) reported a Palestinian Arab boy, born of first-cousin parents, with severe hyperinsulinemic hypoglycemia diagnosed immediately after birth, which failed to respond to medical treatment with either diazoxide or the somatostatin analog, octreotide, and required near-total pancreatectomy to control hypoglycemia. From birth, he had severe vomiting and diarrhea of unknown etiology, which precluded oral feedings for the first 18 weeks of life, but which subsequently remitted. Nestorowicz et al. (1997) noted that they had observed a similar digestive problem in a patient with hyperinsulinism due to mutations in the ABCC8 gene (600509; see also HHF1, 256450) and stated that this patient was clinically indistinguishable from patients with severe hyperinsulinism caused by mutations in the ABCC8 gene.
Congenital hyperinsulinism is a disease where there are abnormally high levels of insulin, a hormone produced by the beta cells of the pancrea s that helps control blood sugar levels. Because of the high levels of insulin, people with this disease have frequent episodes of low blood sugar ( hypoglycemia ) that can even occur after eating. In babies and young children, these episodes are characterized by a lack of energy (lethargy), irritability, or difficulty feeding. Repeated episodes of low blood sugar increase the risk for serious complications such as breathing difficulties, seizures, intellectual disability, vision loss, brain damage, and coma. The severity and onset of these episodes varies, even among members of the same family.
Zeev et al. (2002) reported an Israeli family in which a girl had classic Rett syndrome and her brother had severe neonatal encephalopathy. Leuzzi et al. (2004) reported a 28-month-old boy with neonatal encephalopathy.
Severe neonatal-onset encephalopathy with microcephaly is a rare monogenic disease with epilepsy characterized by neonatal-onset encephalopathy, microcephaly, severe developmental delay or absent development, breathing abnormalities (including central hypoventilation and/or respiratory insufficiency), intractable seizures, abnormal muscle tone and involuntary movements. Early death is usual.
MECP2 -related severe neonatal encephalopathy is a neurological disorder that primarily affects males and causes brain dysfunction (encephalopathy). Affected males have a small head size (microcephaly ), poor muscle tone (hypotonia) in infancy, movement disorders, rigidity, and seizures. Infants with this condition appear normal at birth but then develop severe encephalopathy within the first week of life. These babies experience poor feeding, leading to a failure to gain weight and grow at the expected rate (failure to thrive). Individuals with MECP2 -related severe neonatal encephalopathy have severe to profound intellectual disability.
A coding SNP (Y111H; rs17366743) was confirmed to be associated with diabetes incidence and with higher mean fasting glucose over 28 years of follow-up. In a family-based sample of 640 nondiabetic Caucasian Italians, Menzaghi et al. (2010) measured serum adiponectin isoform concentrations and genotyped 3 ADIPOQ SNPs, rs17300539, rs1501299, and rs6773957.
Study of 42 members from 4 generations revealed a consistent linkage of spherocytosis with 1 particular haplotype generated by the 4 probes that were used. Molecular Genetics In a 28-year-old female with congenital spherocytic hemolytic anemia, Jarolim et al. (1991) identified a missense mutation in the band 3 gene (109270.0003).
A number sign (#) is used with this entry because spherocytosis type 2 (SPH2) is caused by heterozygous mutation in the SPTB gene (182870) on chromosome 14q23. Some patients have been reported with homozygous or compound heterozygous mutations. Description Hereditary spherocytosis refers to a group of heterogeneous disorders that are characterized by the presence of spherical-shaped erythrocytes (spherocytes) on the peripheral blood smear. The disorders are characterized clinically by anemia, jaundice, and splenomegaly, with variable severity. Common complications include cholelithiasis, hemolytic episodes, and aplastic crises (review by Perrotta et al., 2008).
A number sign (#) is used with this entry because hereditary spherocytosis-3 is caused by mutation in the alpha-spectrin gene (SPTA; 182860) on chromosome 1q21. For a general description and a discussion of heterogeneity of spherocytosis, see 182900. Clinical Features Agre et al. (1982) reported 2 daughters, of related but normal parents, who had nearly fatal hemolytic anemia requiring early splenectomy. Both improved strikingly thereafter but spherocytosis persisted. Red cell membranes were at least 50% deficient in spectrin, with band 1 reduced more than band 2. No defect was found in membrane binding of spectrin or in membrane binding sites (ankyrin).
A number sign (#) is used with this entry because of evidence that spherocytosis type 1 is caused by heterozygous, compound heterozygous, or homozygous mutation in the gene encoding ankyrin (ANK1; 612641) on chromosome 8p11. Description Hereditary spherocytosis refers to a group of heterogeneous disorders that are characterized by the presence of spherical-shaped erythrocytes (spherocytes) on the peripheral blood smear. The disorders are characterized clinically by anemia, jaundice, and splenomegaly, with variable severity. Common complications include cholelithiasis, hemolytic episodes, and aplastic crises (review by Perrotta et al., 2008). Elgsaeter et al. (1986) gave an extensive review of the molecular basis of erythrocyte shape with a discussion of the role of spectrin and other proteins such as ankyrin, actin (102630), band 4.1 (130500), and band 3 (109270), all of which is relevant to the understanding of spherocytosis and elliptocytosis (see 611904).
A number sign (#) is used with this entry because hereditary spherocytosis type 5 is caused by mutation in the gene encoding protein 4.2 (EPB42; 177070) on chromosome 15q15. For a general phenotypic description and a discussion of genetic heterogeneity of hereditary spherocytosis, see SPH1 (182900). Clinical Features Hereditary spherocytosis type 5, which has been observed predominantly in Japanese individuals, is an autosomal recessive disorder that results in a hemolytic anemia associated with abnormally shaped, osmotically fragile red blood cells (Bouhassira et al., 1992). Hayashi et al. (1974) described 4 Japanese patients, 3 of whom were sibs, with hereditary spherocytosis and deficiency of protein 4.2. Nozawa et al. (1974) reported a severe case of hereditary spherocytosis in a 6-year-old Japanese girl with protein 4.2 deficiency who showed improvement with splenectomy.
Hereditary spherocytosis is a condition characterized by hemolytic anemia (when red blood cells are destroyed earlier than normal). Signs and symptoms can range from mild to severe and may include pale skin, fatigue, anemia, jaundice, gallstones , and/or enlargement of the spleen . Other symptoms of hemolytic anemia may include feeling that your heart is pounding or racing (palpitations), feeling dizzy, problems concentrating, and headaches. Some people with a severe form of hereditary spherocytosis may have short stature, delayed puberty, and skeletal abnormalities. The condition is caused by mutations in any of several genes, such as the ANK1 , EPB42 , SLC4A1 , SPTA1 , and SPTB genes.
The authors analyzed 2 RLS-associated SNPs, rs12469063 and 2300478, and found a GG/GG risk haplotype (43% vs 25%, p = 0.0095) in 28 RLS and 140 control brain samples.
Two forms were delineated based on age at onset: a juvenile form with onset before age 15 years (66%), and an adult-onset form starting around the third or fourth decade (28%). All affected patients showed characteristic pelvic and shoulder girdle proximal weakness.
A rare subtype of autosomal dominant limb-girdle muscular dystrophy ,with a variable age of onset, characterized by progressive, proximal weakness and wasting of the shoulder and pelvic musculature (with the pelvic girdle, and especially the ileopsoas muscle, being more affected) and frequent association of calf hypertrophy, dysphagia, arachnodactyly with or without finger contractures and/or distal and axial muscle involvement. Additional features include an abnormal gait, exercise intolerance, myalgia, fatigue and respiratory insufficiency. Cardiac conduction defects are typically not observed.
Ku et al. (2007) tested TLR responses of whole blood and individual leukocyte subsets in 28 patients with IRAK4 deficiency and found that only the TLR3 agonist poly(I:C) could induce production of 11 non-IFN cytokines.
IRAK-4 deficiency is a condition that affects the immune system ( primary immunodeficiency ). It causes recurring severe infections by a type of bacteria called pyogenic bacteria. Individuals with IRAK-4 deficiency seem to be particularly susceptible to infections caused by bacteria called Streptococcus pneumoniae . The deficiency is caused by mutations in the IRAK4 gene and is inherited in an autosomal recessive pattern. Treatment may include intravenous immunoglobulin therapy (IVIG), taking antibiotics before an infection develops, and vaccines.
A number sign (#) is used with this entry because recurrent isolated invasive pneumococcal disease (IPD1) can be caused by mutation in the IRAK4 gene (606883). Another form of this disorder (IPD2; 300640) is caused by mutation in the NEMO gene (300248). Protection against IPD has been associated with a coding single-nucleotide polymorphism (SNP) in the TIRAP gene (606252.0001). Description Recurrent invasive pneumococcal disease (IPD) is defined as 2 episodes of IPD occurring at least 1 month apart, whether caused by the same or different serotypes or strains (Ku et al., 2007). Recurrent IPD occurs in at least 2% of patients in most series, making IPD the most important known risk factor for subsequent IPD.
IRAK-4 deficiency is an inherited disorder of the immune system (primary immunodeficiency). This immunodeficiency leads to recurrent infections by a subset of bacteria known as pyogenic bacteria but not by other infectious agents. (Infection with pyogenic bacteria causes the production of pus.) The most common infections in IRAK-4 deficiency are caused by the Streptococcus pneumoniae , Staphylococcus aureus , and Pseudomonas aeruginosa bacteria. Most people with this condition have their first bacterial infection before age 2, and the infections can be life-threatening in infancy and childhood. Infections become less frequent with age. Most people with IRAK-4 deficiency have invasive bacterial infections, which can involve the blood (septicemia), the membrane covering the brain and spinal cord (meningitis), or the joints (leading to inflammation and arthritis).
Interleukin-1 receptor-associated kinase-4 (IRAK-4) deficiency is an immunodeficiency associated with increased susceptibility to invasive infections caused by pyogenic bacteria. Epidemiology It has been described in less than 15 patients from eight families with onset occurring during childhood. Clinical description The most common recurrent infections in these patients were caused by Streptococcus pneumoniae or Staphylococcus aureus , leading to a range of clinical manifestations such as pneumonia, septic arthritis, cellulitis, osteomyelitis, otitis media, meningitis and sinusitis. In contrast, the patients appeared to be resistant to infection caused by most other bacteria, parasites and viruses but fungal infections have been reported. Although routine immunological evaluations generally gave normal results, the patients displayed reduced inflammatory responses and neutropenia during infectious episodes.
Two hundred and thirty-six subjects consisting of 7 pairs concordant for schizophrenia (6 monozygotic, 1 dizygotic), 52 pairs discordant for schizophrenia (20 monozygotic, 32 dizygotic), and 59 demographically balanced normal pairs (28 monozygotic, 31 dizygotic) were drawn from a twin cohort consisting of all same-sex twins born in Finland from 1940 through 1957.
Infantile convulsions and choreoathetosis Other names Paroxysmal kinesigenic dyskinesia and infantile convulsions Infantile convulsions and choreoathetosis is inherited via an autosomal dominant manner Infantile convulsions and choreoathetosis ( ICCA ) syndrome is a neurological genetic disorder with an autosomal dominant mode of inheritance. It is characterized by the association of benign familial infantile epilepsy (BIFE) at age 3–12 months and later in life with paroxysmal kinesigenic choreoathetosis . The ICCA syndrome was first reported in 1997 in four French families from north-western France and provided the first genetic evidence for common mechanisms shared by benign infantile seizures and paroxysmal dyskinesia. [1] The epileptic origin of PKC has long been a matter of debates [2] and PD have been classified as reflex epilepsies. Indeed, attacks of PKC and epileptic seizures have several characteristics in common, they both are paroxysmal in presentation with a tendency to spontaneous remission, and a subset of PKC responds well to anticonvulsants . This genetic disease has been mapped to chromosome 16p -q12. [3] More than 30 families with the clinical characteristics of ICCA syndrome have been described worldwide so far. [4] [5] Contents 1 Presentation 2 Genetics 3 Diagnosis 4 Management 5 References 6 External links Presentation [ edit ] The specific and familial association of BIFE and PKC defines a novel clinical entity : the infantile convulsions and choreoathetosis syndrome.
Infantile Convulsions and paroxysmal ChoreoAthetosis (ICCA) syndrome is a neurological condition characterized by the occurrence of seizures during the first year of life (Benign familial infantile epilepsy ; see this term) and choreoathetotic dyskinetic attacks during childhood or adolescence. Epidemiology This disorder is rare but the exact prevalence is unknown. Clinical description Benign familial infantile epilepsy begins at 3 to 12 months of age with a family history of the same type of seizures. Seizures are afebrile, partial or sometimes generalized, and normally disappear after the first year of life. During childhood or adolescence, affected individuals present with paroxysmal kinesigenic dyskinesia with frequent and recurrent episodic choreathetotic or dystonic movements that last less than 1 minute.
For the equine form of the disease, see Pituitary pars intermedia dysfunction . Cushing's disease Other names Cushing disease, tertiary or secondary hypercortisolism, tertiary or secondary hypercorticism, Itsenko-Cushing disease [1] [2] Specialty Endocrinology Cushing's disease is one cause of Cushing's syndrome characterised by increased secretion of adrenocorticotropic hormone (ACTH) from the anterior pituitary (secondary hypercortisolism ). This is most often as a result of a pituitary adenoma (specifically pituitary basophilism) or due to excess production of hypothalamus CRH ( corticotropin releasing hormone ) (tertiary hypercortisolism/hypercorticism) that stimulates the synthesis of cortisol by the adrenal glands. Pituitary adenomas are responsible for 80% of endogenous Cushing's syndrome, [3] when excluding Cushing's syndrome from exogenously administered corticosteroids . This should not be confused with ectopic Cushing syndrome or exogenous steroid use. [4] Contents 1 Signs and symptoms 1.1 Common 1.2 Less common 2 Diagnosis 2.1 ACTH blood test 2.2 Dexamethasone suppression test 2.3 ACTH stimulation test 2.4 Imaging 2.5 Inferior petrosal sinus sampling 2.6 Urinary free cortisol test 2.7 Late night (midnight) salivary cortisol test 3 Treatment 4 Epidemiology 5 History 6 References 7 External links Signs and symptoms [ edit ] The symptoms of Cushing's disease are similar to those seen in other causes of Cushing's syndrome . [5] Patients with Cushing's disease usually present with one or more signs and symptoms secondary to the presence of excess cortisol or ACTH . [6] Although uncommon, some patients with Cushing's disease have large pituitary tumors (macroadenomas).
ACTH-secreting pituitary adenoma is a condition characterized by elevated levels of a hormone called cortisol secreted by a tumor in the pituitary gland. It is part of a group of diseases that cause Cushing’s syndrome , characterized by signs and symptoms that may include weight gain around the trunk and in the face, stretch marks, easy bruising, a hump on the upper back, muscle weakness, tiredness, thin bones that are prone to fracture (osteoporosis), mood disorders and memory problems, as well as an increased risk of infections, high blood pressure and diabetes . Women may have irregular menses and a lot of hair in the body (hirsutism). It occurs when a benign pituitary tumor (adenoma) or pituitary hyperplasia causes the adrenal glands to produce large amounts of cortisol. Some cases are caused by somatic mutations in the AIP and the GNAS genes.
Overview Cushing syndrome happens when the body has too much of the hormone cortisol for a long time. This can result from the body making too much cortisol, or from taking medicines called glucocorticoids, which affect the body the same way as cortisol. Too much cortisol can cause some of the main symptoms of Cushing syndrome — a fatty hump between the shoulders, a rounded face, and pink or purple stretch marks on the skin. Cushing syndrome also can cause high blood pressure or bone loss. Sometimes, it can cause type 2 diabetes. Treatments for Cushing syndrome can lower the body's cortisol levels and improve symptoms.
Cushing disease (CD) is the most common cause of endogenous Cushing syndrome (CS; see this term) and is due to pituitary chronic over-secretion of ACTH by a pituitary corticotroph adenoma. Epidemiology Exact prevalence is unknown. Prevalence of endogenous CS is estimated at around 1/26,000, with CD representing more than two-thirds of all cases. Recent data suggests that mild CD is more common than previously thought. Clinical description The female-to-male ratio of CD is 4-5:1, except in prepubertal patients, in which a strong male predominance is observed. The peak incidence is at 25-40 years of age. The disease manifests with signs of CS (truncal and facial obesity and signs of hypercatabolism) as well as skin hyperpigmentation and/or neurological complications in some cases of corticotroph macro-adenoma.
More commonly it presents during traumatic bleeding or surgical procedures. [2] [3] Most cases (60%) of dysfibrinogenemia are asymptomatic, but 28% exhibit hemorrhaging similar to that described above while 20% exhibit thrombosis (i.e. excessive clotting). [3] Causes [ edit ] The disorders associated with Factor I deficiency are generally inherited , [2] [3] although certain liver diseases can also affect fibrinogen levels and function (e.g. cirrhosis ). [8] Afibrinogenemia is a recessive inherited disorder, where both parents must be carriers. [2] Hypofibrinogenemia and dysfibrinogenemia can be dominant (i.e. only one parent needs to be a carrier) or recessive. [2] The origin of the disorders is traced back to three possible genes: FGA, FGB, or FGG.
Complement factor I deficiency is a disorder that affects the immune system. People with this condition are prone to recurrent infections, including infections of the upper respiratory tract, ears, skin, and urinary tract. They may also contract more serious infections such as pneumonia, meningitis, and sepsis , which may be life-threatening. Some people with complement factor I deficiency have a kidney disorder called glomerulonephritis with isolated C3 deposits. Complement factor I deficiency can also be associated with autoimmune disorders such as rheumatoid arthritis or systemic lupus erythematosus (SLE).
A number sign (#) is used with this entry because complement factor I ('eye') deficiency (CFID) is caused by homozygous or compound heterozygous mutation in the gene encoding complement factor I (CFI; 217030) on chromosome 4q25. Description Hereditary deficiency of complement factor I is associated with a propensity to pyogenic infection and follows an autosomal recessive pattern of inheritance (Vyse et al., 1996). See also complement factor H deficiency (609814), which shows overlapping clinical features. Clinical Features Alper et al. (1970, 1970) and Abramson et al. (1971) reported a patient with increased susceptibility to infection and accelerated catabolism of C3 due to deficiency of the C3 inactivator. Alper et al. (1972) demonstrated that the C3 inactivator also acts as an inhibitor in the alternative complement pathway.
Immunodeficiency with factor I anomaly is a rare, genetic, primary immunodeficiency disease characterized by increased susceptibility to recurrent, usually severe, infections (particularly by Neisseria meningitidis , Haemophilus influenzae and Streptococcus pneumonia ), typically manifesting as otitis, sinusitis, bronchitis, pneumonia, and/or meningitis. Autoimmune disease (e.g. systemic lupus erythematosus, glomerulonephritis) and atypical hemolytic uremic syndrome may be associated. Laboratory serum analysis reveals, in addition to diminished or undetectable complement factor I, variably decreased complement C3, complement factor B and complement factor H.