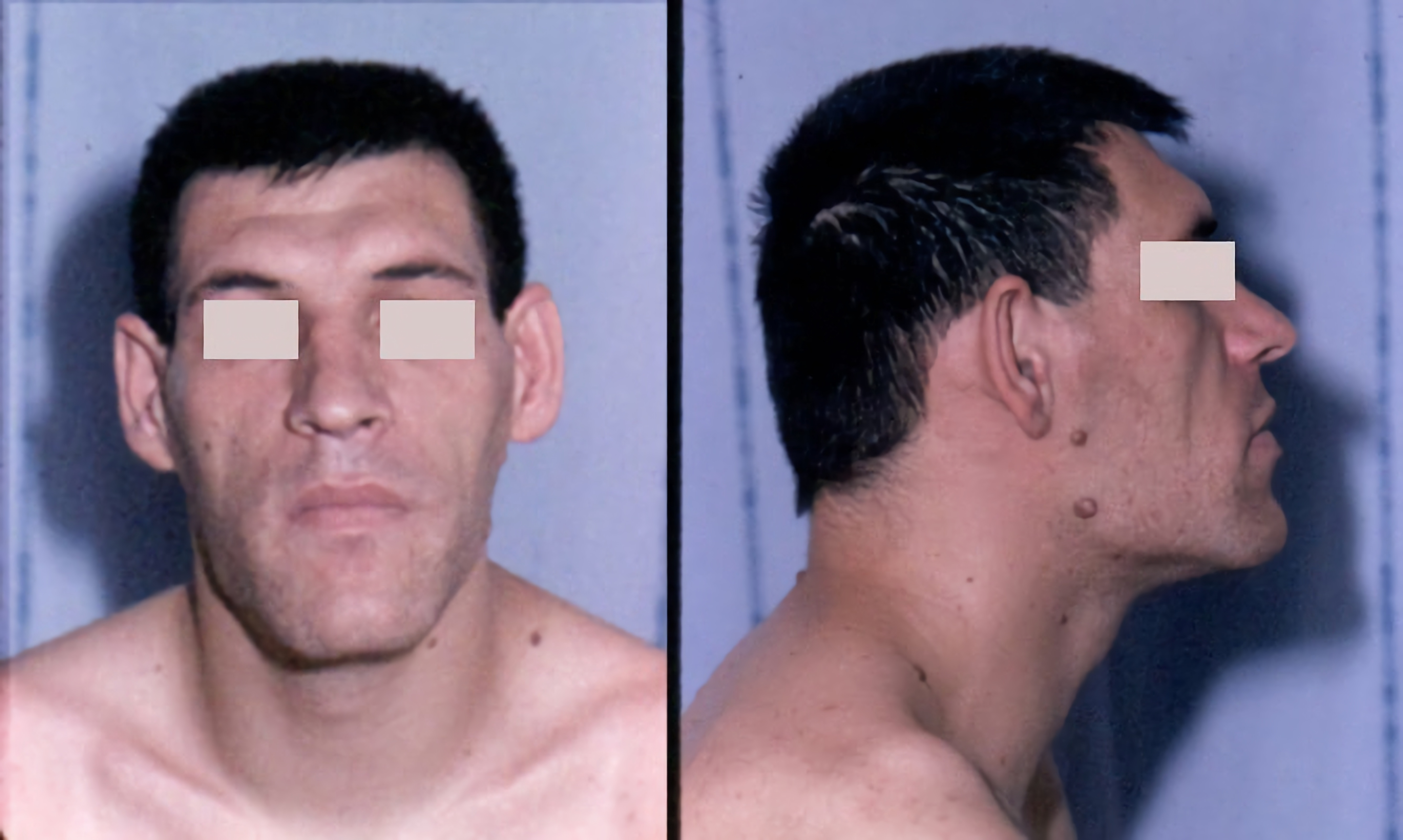
Clinical description NARP (Neuropathy, Ataxia, and Retinitis Pigmentosa) syndrome is characterized by a great phenotypic variability and habitually clearly manifests in young adulthood. ... The m.8993T>G mutation is also present in 8-10% of patients with Leigh syndrome when mutated mitochondrial DNA is > 90%. ... Differential diagnosis Differential diagnosis includes maternally inherited Leigh syndrome (MILS, like NARP syndrome part of the same group of disorders of mitochondrial oxidative phosphorylation), Refsum disease, Cockayne syndrome, abetalipoproteinemia, Usher syndrome (see these terms), neurological complication of lipidosis or rare cases of spinocerebellar disorder/ataxia. ... Preimplantation genetic diagnosis is available for affected couples in few centers. Genetic counseling NARP syndrome is a maternally inherited syndrome and women can transmit to all their offspring. ... Agents to avoid include Sodium Valproate, barbiturates and anesthesia (in general) as well as dichloroacetate. Prognosis As NARP syndrome is progressive, patients may become increasingly dependent of others.
A number sign (#) is used with this entry because of evidence that NARP syndrome is caused by mutation in the gene encoding subunit 6 of mitochondrial H(+)-ATPase (MTATP6; 516060). ... Lopez-Gallardo et al. (2009) reported a 40-year-old man with NARP syndrome. He had delayed development, psychomotor retardation, and irritability in childhood, and later developed other neurologic signs, including hearing loss, blindness due to optic atrophy and retinitis pigmentosa, ataxia, and clonic spasms.
Antioxidants play a role in improving the oxidative phosphorylation that is otherwise impaired. [11] Prognosis [ edit ] The severity and prognosis vary with the type of mutation involved. [12] See also [ edit ] Leigh's disease References [ edit ] ^ "Maternally inherited Leigh syndrome and NARP syndrome" . rarediseases.org . ... "Cone and rod dysfunction in the NARP syndrome" . The British Journal of Ophthalmology . 83 (2): 190–3. doi : 10.1136/bjo.83.2.190 . ... PMID 10396197 . ^ Keränen, T; Kuusisto, H (Sep 2006). "NARP syndrome and adult-onset generalised seizures" . ... Mitochondrial DNA-Associated Leigh Syndrome and NARP . University of Washington, Seattle. ... External links [ edit ] Classification D ICD - 10 : G31.8 ICD - 9-CM : 277.87 OMIM : 551500 MeSH : C537396 DiseasesDB : 34335 External resources Orphanet : 644 v t e Mitochondrial diseases Carbohydrate metabolism PCD PDHA Primarily nervous system Leigh disease LHON NARP Myopathies KSS Mitochondrial encephalomyopathy MELAS MERRF PEO No primary system DAD MNGIE Pearson syndrome Chromosomal OPA1 Kjer's optic neuropathy SARS2 HUPRA syndrome TIMM8A Mohr–Tranebjærg syndrome see also mitochondrial proteins
This disorder is probably less common than a similar but more severe condition, Leigh syndrome, which affects about 1 in 40,000 people. ... When this mutation is present in a higher percentage of a person's mitochondria—more than 90 percent to 95 percent—it usually causes a more severe condition known as maternally inherited Leigh syndrome. Because these two conditions result from the same genetic changes and can occur in different members of a single family, and because some individuals with MT-ATP6 gene mutations have related signs and symptoms that do not follow the specific patterns of these conditions, researchers believe that the conditions may be part of a spectrum of overlapping features rather than two distinct syndromes.
Neuropathy ataxia retinitis pigmentosa (NARP) syndrome is characterized by a variety of signs and symptoms that mainly affect the nervous system. ... Mutations in the MT-ATP6 gene cause NARP syndrome. This gene is located within mitochondrial DNA (mtDNA). Most individuals with NARP have a specific MT-ATP6 mutation in 70 percent to 90 percent of their mitochondria. NARP syndrome is inherited from the mother (maternal inheritance) because only females pass mitochondrial DNA to their children.
Fechtner syndrome Other names Alport syndrome with leukocyte inclusions and macrothrombocytopenia Fechtner syndrome is inherited in an autosomal dominant manner. Fechtner syndrome is a variant of Alport syndrome characterized by leukocyte inclusions, macrothrombocytopenia , [1] thrombocytopenia , nephritis , and sensorineural hearing loss . [2] Some patients may also develop cataracts. [3] References [ edit ] ^ cause by mutation in the MYH9 gene on chromosome 22q11 AbstractThis study reports a family comprising four generations in whom nephritis, deafness, congenital cataracts, macrothrombocytopenia, and leukocyte inclusions were observed in varying combinations in eight of 17 members. ... Renal disease ranged from Peterson LC, Rao KV, Crosson JT, White JG (February 1985). "Fechtner syndrome--a variant of Alport's syndrome with leukocyte inclusions and macrothrombocytopenia" . ... Hereditary Hearing Loss and Its Syndromes . Oxford University Press USA. p. 127.
Combined clinical phenotype caused by each gene involved in a chromosomal abnormality A contiguous gene syndrome (CGS), also known as a contiguous gene deletion syndrome is a clinical phenotype caused by a chromosomal abnormality, such as a deletion or duplication that removes several genes lying in close proximity to one another on the chromosome. ... While it can be caused by deleted material on a chromosome, it is not, strictly speaking, the same entity as a segmental aneuploidy syndrome. A segmental aneuploidy syndrome is a subtype of CGS that regularly recur, usually due to non-allelic homologous recombination between low copy repeats in the region. [1] Most CGS involve the X chromosome and affect male individuals. [2] One of the earliest and most famous examples of a CGS involves a male patient with Duchenne muscular dystrophy (DMD), chronic granulomatous disease (CGD), retinitis pigmentosa and intellectual disability . ... In addition to the previously described CGS that occur on the X chromosome, two other common syndromes are Langer-Giedion syndrome (caused by deletions of TRPS1 and EXT1 on 8q24 and WAGR syndrome (caused by deletions on 11q13 encompassing PAX6 and WT1 .) [1] References [ edit ] ^ a b c Strachan, Tom; Read, Andrew. ... "Molecular Cytogenetics of Contiguous Gene Syndromes: Mechanisms and Consequences of Gene Dosage Imbalance".
Frontal bossing may be seen in a few rare medical syndromes such as acromegaly – a chronic medical disorder in which the anterior pituitary gland produces excess growth hormone (GH). [2] Frontal bossing may also occur in diseases resulting in chronic anemia, where there is increased hematopoiesis and enlargement of the medullary cavities of the skull . [1] Associated medical disorders [ edit ] Rickets [3] Achondroplasia Acromegaly Basal cell nevus syndrome Congenital syphilis Cleidocranial dysostosis Crouzon syndrome Cryopyrin-Associated Periodic Syndrome (CAPS – PFS) [4] Ectodermal dysplasia Extramedullary hematopoiesis Fragile X syndrome Hurler syndrome Osteopathia Striata with Cranial Sclerosis Pfeiffer syndrome Rubinstein-Taybi syndrome Russell-Silver syndrome (Russell-Silver dwarf) Thanatophoric dysplasia Talfan syndrome Trimethadione (antiseizure drug) use during pregnancy Beta-thalassemia (due to expansion of bone marrow secondary to increased hematopoiesis ) [5] Hallermann-Streiff syndrome References [ edit ] ^ a b Dennis, Mark; Bowen, William Talbot; Cho, Lucy (2012).
Muckle-Wells syndrome is a disorder characterized by periodic episodes of skin rash, fever, and joint pain. Progressive hearing loss and kidney damage also occur in this disorder. People with Muckle-Wells syndrome have recurrent "flare-ups" that begin during infancy or early childhood. ... Abnormal deposits of a protein called amyloid (amyloidosis) cause progressive kidney damage in about one-third of people with Muckle-Wells syndrome; these deposits may also damage other organs. ... Causes Mutations in the NLRP3 gene (also known as CIAS1 ) cause Muckle-Wells syndrome. The NLRP3 gene provides instructions for making a protein called cryopyrin. ... Researchers believe that NLRP3 gene mutations that cause Muckle-Wells syndrome result in a hyperactive cryopyrin protein and an inappropriate inflammatory response.
Muckle–Wells syndrome Other names Urticaria-deafness-amyloidosis syndrome ( UDA ), [1] This condition is inherited in an autosomal dominant manner Specialty Dermatology Muckle–Wells syndrome ( MWS ) is a rare autosomal dominant disease which causes sensorineural deafness and recurrent hives , and can lead to amyloidosis . ... As a result, MWS is considered a type of periodic fever syndrome . MWS is caused by a defect in the CIAS1 gene which creates the protein cryopyrin . MWS is closely related to two other syndromes, familial cold urticaria and neonatal onset multisystem inflammatory disease —in fact, all three are related to mutations in the same gene and subsumed under the term cryopyrin-associated periodic syndromes (CAPS). ... "Hearing improvement in a patient with variant Muckle‐Wells syndrome in response to interleukin 1 receptor antagonism" . ... "Urticaria, deafness, and amyloidosis: a new heredo-familial syndrome". The Quarterly Journal of Medicine . 31 : 235–48.
Muckle-Wells syndrome (MWS) is an intermediate form of cryopyrin-associated periodic syndrome (CAPS; see this term) and is characterized by recurrent fever (with malaise and chills), recurrent urticaria-like skin rash, sensorineural deafness, general signs of inflammation (eye redness, headaches, arthralgia/myalgia) and potentially life-threatening secondary amyloidosis (AA type). ... Mutations in this gene may also cause two additional phenotypes of CAPS: familial cold urticaria (FCAS) and CINCA syndrome (see these terms), Patients carrying identical amino acid substitution may present with distinctly different clinical subtypes, suggesting that additional genetic and/or environmental modifying factors are important in disease expression. ... Some patients with a classical phenotype of MWS, FCAS or CINCA syndrome may not have mutations in NLRP3 .
A number sign (#) is used with this entry because of evidence that Muckle-Wells syndrome is caused by heterozygous mutation in the gene encoding cryopyrin (NLRP3; 606416) on chromosome 1q44. ... See also familial cold-induced autoinflammatory syndrome-1 (FCAS1, CAPS1; 120100), an allelic disorder with overlapping clinical features. ... Gerbig et al. (1998) stated that about 100 cases of the urticaria-deafness-amyloidosis syndrome had been reported since the description of the syndrome in 9 members of a Derbyshire family by Muckle and Wells (1962). ... In his thirties and forties, he had recurrent abdominal pain, profuse diarrhea, and nephrotic syndrome requiring peritoneal dialysis. ... Molecular Genetics In a family with Muckle-Wells syndrome, Hoffman et al. (2001) found a mutation in the NLRP3 gene (606416.0004).
Morgagni Stewart Morel syndrome Other names Hyperostosis frontalis interna,' Metabolic craniopathy Morgagni Stewart Morel syndrome is inherited in an X-linked recessive manner(or autosomal dominant). [1] Specialty Endocrinology Morgagni-Stewart-Morel syndrome is a condition with a wide range of associated endocrine problems including: diabetes mellitus , diabetes insipidus , and hyperparathyroidism . [2] Other signs and symptoms include headaches , vertigo , hirsutism , menstrual disorder , galactorrhoea , obesity , depression , and seizures . [2] Thickening of the inner table of the frontal part of the skull a usually benign condition known as hyperostosis frontalis interna . [2] [3] The syndrome was first described in 1765. [3] It is named after the Italian anatomist and pathologist Giovanni Battista Morgagni , the British neurologist Roy Mackenzie Stewart , and the Swiss psychiatrist Ferdinand Morel . ... Seizures and headaches associated with hyperostosis frontalis interna (HFI) are treated with standard medications. [6] References [ edit ] ^ INSERM US14. "Morgagni Stewart Morel syndrome" . Orphanet . Retrieved 1 November 2017 . ^ a b c Nallegowda M, Singh U, Khanna M, Yadav SL, Choudhary AR, Thakar A (March 2005). "Morgagni Stewart Morel syndrome—additional features" . Neurol India . 53 (1): 117–9. doi : 10.4103/0028-3886.15078 . hdl : 1807/7758 . ... Retrieved 2018-04-17 . ^ "Morgagni-Stewart-Morel syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . Retrieved 2018-04-17 . ^ "Morgagni-Stewart-Morel syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov .
Morgagni-Stewart-Morel (MSM) syndrome is a disorder characterized by thickening of the frontal bone of the skull (hyperostosis frontalis interna), as well as obesity and excessive hair growth (hypertrichosis). ... The cause of Morgagni-Stewart-Morel syndrome is not fully understood. Some instances of dominant inheritance have been reported, but whether it is autosomal dominant or X-linked dominant is not known.
A rare cranial malformation characterized by hyperostosis frontalis interna, variably associated with metabolic and endocrine disorders (such as obesity, diabetes mellitus, and hirsutism, among others). Compression by calvarial thickening may lead to cerebral atrophy and present with cognitive impairment, neuropsychiatric symptoms, headaches, and epilepsy. The condition predominantly affects women.
Hyperostosis frontalis interna Hyperostosis frontalis interna in a 74-year-old woman Specialty Radiology Hyperostosis frontalis interna is a common, benign thickening of the inner side of the frontal bone of the skull . It is found predominantly in women after menopause and is usually asymptomatic. Mostly frequently it is found as an incidental finding discovered during an X-ray or CT scan of the skull. Additional images [ edit ] Hyperostosis frontalis at CT References [ edit ] She R, Szakacs J (2004). "Hyperostosis frontalis interna: case report and review of literature".
Since hyperprolactinemia was found in many of these cases, the authors suggested that this and other features of the syndrome such as hirsutism, diabetes, and menstrual troubles may be related to hyperprolactinemia.
Osteoporosis-pseudoglioma syndrome is a rare condition characterized by severe thinning of the bones (osteoporosis) and eye abnormalities that lead to vision loss. ... Frequency Osteoporosis-pseudoglioma syndrome is a rare disorder that occurs in approximately 1 in 2 million people. Causes Osteoporosis-pseudoglioma syndrome is caused by mutations in the LRP5 gene. ... LRP5 gene mutations that cause osteoporosis-pseudoglioma syndrome prevent cells from making any LRP5 protein or lead to a protein that cannot function. Loss of this protein's function disrupts the chemical signaling pathways that are needed for the formation of bone and for normal retinal development, leading to the bone and eye abnormalities characteristic of osteoporosis-pseudoglioma syndrome. Learn more about the gene associated with Osteoporosis-pseudoglioma syndrome LRP5 Inheritance Pattern Osteoporosis-pseudoglioma syndrome is inherited in an autosomal recessive pattern , which means both copies of the LRP5 gene in each cell have mutations.
A number sign (#) is used with this entry because osteoporosis-pseudoglioma syndrome is caused by homozygous or compound heterozygous mutation in the gene encoding low density lipoprotein receptor-related protein-5 (LRP5; 603506) on chromosome 11q13. ... Robinow (1985), Brude (1986), and Superti-Furga et al. (1986) suggested that this is really the osteoporosis-pseudoglioma syndrome. Two obligate heterozygotes in the family described by Superti-Furga et al. (1986) had had 3 fractures each and had osteoporosis, suggesting the possibility of clinical expression of this recessive gene in heterozygotes (Superti-Furga, 1989). Frontali and Dallapiccola (1986) likewise concluded that Beighton's ocular osteogenesis imperfecta is osteoporosis-pseudoglioma syndrome, and Beighton (1986) acknowledged the diagnosis. ... Inheritance The osteoporosis-pseudoglioma syndrome is inherited as an autosomal recessive trait (Neuhauser et al., 1976).
Osteoporosis pseudoglioma syndrome is a very rare autosomal recessive disorder characterized by congenital or infancy-onset blindness and severe juvenile-onset osteoporosis and spontaneous fractures.
The features were identical to those of the cases reported by Ramon et al. (1967) except that 3 of the 4 patients also had juvenile rheumatoid arthritis, which de Pina-Neto et al. (1986) suggested should be considered a part of the syndrome. De Pina-Neto et al. (1998) provided information on the clinical evolution of the disorder in the Brazilian family. ... Parkin and Law (2001) reported follow-up of 2 sibs with Ramon syndrome, originally described by Pridmore et al. (1992). ... In addition, both sibs had bilateral anterior chamber eye anomalies (Axenfeld anomaly), not previously described in Ramon syndrome. The authors suggested that ocular abnormalities may be another feature of this syndrome. Some of the features of Ramon syndrome are found in patients treated with phenytoin. Gingival hyperplasia also occurs in the Rutherfurd syndrome (180900), the Laband syndrome (135500), and the Jones syndrome (135550).
A rare, genetic, primary bone dysplasia syndrome characterized by bilateral, painless swelling of the face extending from the mandible to the inferior orbital margins (cherubism), epilepsy, gingival fibromatosis (possibly obscuring teeth), and intellectual disability.
Description The Martinez-Frias syndrome is characterized by pancreatic hypoplasia, intestinal atresia, and gallbladder aplasia or hypoplasia, with or without tracheoesophageal fistula. There is considerable phenotypic overlap between Martinez-Frias syndrome and Mitchell-Riley syndrome (MTCHRS; 615710), the latter being characterized by neonatal diabetes in addition to the features of the Martinez-Frias syndrome, but without tracheoesophageal fistula (Smith et al., 2010). Clinical Features Martinez-Frias et al. (1992) reported a seemingly distinct autosomal recessive syndrome in a brother and sister born to consanguineous Spanish Gypsy parents. ... Anneren et al. (1998) described a brother and sister, born of healthy consanguineous parents, with a multiple congenital anomalies (MCA) syndrome that bore both similarities and differences to the syndrome described by Martinez-Frias et al. (1992). ... Molecular Genetics Exclusion Studies In the parents of a deceased male infant with Martinez-Frias syndrome, originally described by Gentile and Fiorente (1999), Smith et al. (2010) analyzed the candidate gene RFX6 (612659) and did not find any mutations.
Hutchinson-Gilford progeria syndrome is a genetic condition characterized by the dramatic, rapid appearance of aging beginning in childhood. ... People with Hutchinson-Gilford progeria syndrome experience severe hardening of the arteries (arteriosclerosis ) beginning in childhood. ... Causes Mutations in the LMNA gene cause Hutchinson-Gilford progeria syndrome. The LMNA gene provides instructions for making a protein called lamin A. ... Mutations that cause Hutchinson-Gilford progeria syndrome result in the production of an abnormal version of the lamin A protein. ... Researchers are working to determine how these changes lead to the characteristic features of Hutchinson-Gilford progeria syndrome. Learn more about the gene associated with Hutchinson-Gilford progeria syndrome LMNA Inheritance Pattern Hutchinson-Gilford progeria syndrome is considered an autosomal dominant condition, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Summary Clinical characteristics. Hutchinson-Gilford progeria syndrome (HGPS) is characterized by clinical features that typically develop in childhood and resemble some features of accelerated aging. ... Non-progeroid laminopathies (see Differential Diagnosis) 5. Non-laminopathy progeroid syndromes (see Differential Diagnosis) The diagnosis of classic genotype HGPS is established in a proband with the above Suggestive Findings and a heterozygous c.1824C>T pathogenic variant in LMNA identified on molecular genetic testing (see Table 1). ... Clinical Description Classic and nonclassic genotype Hutchinson-Gilford progeria syndrome (HGPS) are characterized by clinical features that develop in childhood and resemble some features of accelerated aging. ... Nomenclature HGPS is also referred to as the Hutchinson-Gilford syndrome or progeria. Prevalence The prevalence of children with HGPS per total population is one in 20 million [Gordon et al 2014]. ... Differential Diagnosis Non-laminopathy progeroid syndromes. Other syndromes that include some features of premature aging: Neonatal progeroid syndrome (Wiedemann-Rautenstrauch syndrome) (OMIM 264090) Acrogeria (OMIM 201200) Cockayne syndrome Hallermann-Streiff syndrome (OMIM 234100) Gerodermia osteodysplastica (OMIM 231070) Berardinelli-Seip congenital lipodystrophy (congenital generalized lipodystrophy) Petty-Laxova-Weidemann progeroid syndrome (OMIM 612289) Ehlers-Danlos syndrome, progeroid form (OMIM 130070) Werner syndrome Mandibuloacral dysplasia (see Genetically Related Disorders) (OMIM 248370) Nestor-Guillermo syndrome (OMIM 614008) Penttinen Syndrome (OMIM 601812) POLR3A -related Wiedemann-Rautenstrauch syndrome (see POLR3-Related Leukodystrophy, Wambach et al [2018]) PYCR1 -related Wiedemann-Rautenstrauch-like syndrome [Lessel et al 2018] Management Evaluations Following Initial Diagnosis To establish the extent of disease and needs in an individual diagnosed with Hutchinson-Gilford progeria syndrome (HGPS), the following evaluations are recommended if they have not already been completed: Weight and height plotted on standard growth charts to evaluate growth over time Electrocardiogram (ECG) and echocardiogram Carotid artery duplex scans to evaluate size of the lumen and intimal thickness in order to establish baseline vascular status MRI/MRA of the brain and neck Skeletal x-rays to evaluate for characteristic findings: acroosteolysis, clavicular resorption, coxa valga, and extraskeletal soft tissue calcifications [Cleveland et al 2012] Orthopedic evaluation for progressive coxa valga and/or avascular necrosis Dual-energy x-ray absorptiometry (DXA) to assess bone mineral density.
The designation Hutchinson-Gilford progeria syndrome appears to have been first used by DeBusk (1972). ... These individuals had a more severe phenotype than those with RECQL2-associated Werner syndrome. Although Chen et al. (2003) designated these patients as having 'atypical Werner syndrome,' Hegele (2003) suggested that the patients more likely had late-onset Hutchinson-Gilford progeria syndrome. Hegele (2003) reviewed the clinical features of the 4 patients with LMNA mutations reported by Chen et al. (2003), and stated that the designation of 'atypical Werner syndrome' appeared somewhat insecure. He noted that the comparatively young ages of onset in the patients with mutant LMNA would be just as consistent with late-onset HGPS as with early-onset Werner syndrome. ... Hegele (2003) suggested that genomic DNA analysis can help draw a diagnostic line that clarifies potential overlap between older patients with Hutchinson-Gilford syndrome and younger patients with Werner syndrome, and that therapies may depend on precise molecular classification. ... Kane et al. (2013) reported a family in which 5 individuals had a progeroid syndrome with prominent cutaneous and cardiovascular manifestations.
Overview Progeria (pro-JEER-e-uh), also known as Hutchinson-Gilford progeria syndrome, is an extremely rare, progressive genetic disorder. ... In most cases, the rare gene change that causes progeria happens by chance. Other similar syndromes There are other syndromes that may include problems with progerin-like proteins. These conditions are called progeroid syndromes. The changed genes that cause these syndromes are passed down in families. They cause rapid aging and a shortened life span: Wiedemann-Rautenstrauch syndrome, also known as neonatal progeroid syndrome, starts in the womb, with symptoms of aging apparent at birth. Werner syndrome, also known as adult progeria, begins in the teen years or early adulthood.
Progeria leads to extreme premature aging and affects many different body systems. The symptoms begin within a year of life with poor growth and weight gain. Children with progeria have a characteristic facial appearance with a large head, small mouth and chin, narrow nose and large eyes. Other symptoms include baldness, loss of fat under the skin, and dental and joint abnormalities. They also often have symptoms typically seen in much older people including joint stiffness, hip dislocations and severe, progressive heart disease.
Hutchinson-Gilford progeria syndrome is a rare, fatal, autosomal dominant and premature aging disease, beginning in childhood and characterized by growth reduction, failure to thrive, a typical facial appearance (prominent forehead, protuberant eyes, thin nose with a beaked tip, thin lips, micrognathia and protruding ears) and distinct dermatologic features (generalized alopecia, aged-looking skin, sclerotic and dimpled skin over the abdomen and extremities, prominent cutaneous vasculature, dyspigmentation, nail hypoplasia and loss of subcutaneous fat).
Jugular foramen syndrome Human brain(normal) inferior view showing cranial nerves Jugular foramen syndrome , or Vernet's syndrome , is characterized by paresis of the glossopharyngeal , vagal , and accessory (with or without the hypoglossal ) nerves. [1] [2] Contents 1 Symptoms 2 Causes 3 Diagnosis 4 References Symptoms [ edit ] Symptoms of this syndrome are consequences of this paresis. As such, in an affected patient, you may find: dysphonia/hoarseness soft palate dropping deviation of the uvula towards the normal side dysphagia loss of sensory function from the posterior 1/3 of the tongue (CN IX) decrease in the parotid gland secretion (CN IX) loss of gag reflex sternocleidomastoid and trapezius muscles paresis (CN XI) Causes [ edit ] Glomus tumors (most frequently) Meningiomas Schwannomas ( Acoustic neuroma ) Metastatic tumors located at the cerebellopontine angle Trauma Fracture of occipital bone Infections Cholesteatoma (very rare) Obstruction of the jugular foramen due to bone diseases Nasopharyngeal carcinoma spreading into the parapharyngeal space involving the ninth, tenth, and eleventh cranial nerves Diagnosis [ edit ] Gadolinium enhanced mri for vestibular schwannoma mri and biopsy for nasopharyngeal carcinoma based on nerve palsies NCCT for occipital bone fracture References [ edit ] ^ Erol FS, Kaplan M, Kavakli A, Ozveren MF.Jugular foramen syndrome caused by choleastatoma. Clin Neurol Neurosurg. 2005 Jun;107(4):342-6. ^ Quinones-Hinojosa, Alfredo, ed. (2012). ... ISBN 9781455723287 . v t e Cranial nerve disease Olfactory Optic Oculomotor Oculomotor nerve palsy Trochlear Trochlear nerve palsy Trigeminal Trigeminal neuralgia Abducens Abducens nerve palsy Facial Central facial palsy Facial nerve paralysis Bell's palsy Vestibulocochlear Glossopharyngeal Vagus Accessory Accessory nerve disorder Hypoglossal Combined syndromes Bulbar palsy Jugular foramen syndrome Cavernous sinus thrombosis
Absent pulmonary valve syndrome Other names Congenital absence of the pulmonary valve Specialty Cardiology Absent pulmonary valve syndrome is a congenital heart defect that occurs when the flaps of the pulmonary valve do not develop or are severely underdeveloped ( hypoplasia ) resulting in aneurysms (dilation) of the pulmonary arteries and softening of the trachea and bronchi ( tracheobronchomalacia ). ... "The Lecompte Maneuver for Relief of Airway Compression in Absent Pulmonary Valve Syndrome". The Annals of Thoracic Surgery . 81 (5): 1802–1807. doi : 10.1016/j.athoracsur.2005.12.001 . ... PMID 16631676 . v t e Congenital heart defects Heart septal defect Aortopulmonary septal defect Double outlet right ventricle Taussig–Bing syndrome Transposition of the great vessels dextro levo Persistent truncus arteriosus Aortopulmonary window Atrial septal defect Sinus venosus atrial septal defect Lutembacher's syndrome Ventricular septal defect Tetralogy of Fallot Atrioventricular septal defect Ostium primum Consequences Cardiac shunt Cyanotic heart disease Eisenmenger syndrome Valvular heart disease Right pulmonary valves stenosis insufficiency absence tricuspid valves stenosis atresia Ebstein's anomaly Left aortic valves stenosis insufficiency bicuspid mitral valves stenosis regurgitation Other Underdeveloped heart chambers right left Uhl anomaly Dextrocardia Levocardia Cor triatriatum Crisscross heart Brugada syndrome Coronary artery anomaly Anomalous aortic origin of a coronary artery Ventricular inversion This article about a medical condition affecting the circulatory system is a stub .
It usually occurs in association with additional cardiovascular malformations such as teralogy of fallot or ventricular septal defect, or can occur as part of a syndrome (e.g. 22q11.2 deletion syndrome).
Jervell and Lange-Nielsen syndrome is a condition that causes profound hearing loss from birth and a disruption of the heart's normal rhythm (arrhythmia). This disorder is a form of long QT syndrome, which is a heart condition that causes the heart (cardiac) muscle to take longer than usual to recharge between beats. ... Frequency Jervell and Lange-Nielsen syndrome is uncommon; it affects an estimated 1.6 to 6 per 1 million people worldwide. ... Causes Jervell and Lange-Nielsen syndrome is caused by mutations in the KCNE1 and KCNQ1 genes. ... About 90 percent of cases of Jervell and Lange-Nielsen syndrome are caused by mutations in the KCNQ1 gene; KCNE1 mutations are responsible for the remaining cases.
Ascher's syndrome Other names Laffer-Ascher Syndrome Ascher's syndrome , is a rare disorder first described in 1920. [1] It is characterized by repeated episodes of lip and eyelid edema and occasionally euthyroid goiter . The syndrome generally occurs within the first 20 years of life. [2] About 100 cases had been described by 1998. [3] Contents 1 Signs and Symptoms 2 Diagnosis 3 Treatment 4 References 5 External links Signs and Symptoms [ edit ] Blepharochalasis : Recurrent episodes of swelling cause stretching and atrophy of the upper eyelid skin. ... You can help by adding to it . ( April 2018 ) Treatment [ edit ] Cosmetic surgery is generally the treatment of choice. [4] References [ edit ] ^ a b c Gorlin RJ, Pindborg JJ, CohenMM.Syndromes of the head and neck, 4th ed.New York:McGraw-Hill, 1976: 500-501. ^ a b Sanchez MR, Lee M, Moy JA et al. Ascher syndrome: a mimicker of acquired angioedema. ... "Double lip in a patient with Ascher's syndrome". European Journal of Plastic Surgery . 21 (7): 370–373. doi : 10.1007/s002380050120 . ^ Atzeni M, et al. Surgical correction and MR imaging of double lip in Ascher syndrome: record of a case and a review of the literature.
Ascher syndrome is a rare condition characterized by a combination of episodic edemea or swelling of the eyelids (blepharochalasia), double lip, and nontoxic thyroid enlargement ( goiter ).
A very rare syndrome characterized by a combination of blepharochalasis, double lip, and non-toxic thyroid enlargement (seen in 10-50% of cases), although the occurrence of all three signs at presentation is uncommon.
Franceschetti (1955) described the syndrome in father and daughter. Sagging eyelids and double upper lip are features. ... The authors suggested that the features in this patient constituted a syndrome that was distinct from Ascher syndrome.
Wilson studied 14 males from three successive generations that presented hypogonadism, mental retardation, gynecomastia , and short stature, among other symptoms. [4] Eventually, this disorder was ruled distinct from a syndrome presented by Prader and Willi ( Prader-Willi syndrome ) because of its mode of inheritance, gynecomastia, and the presence of small hands and feet. [5] However, there is some speculation that this syndrome is in the same spectrum as the Cornelia de Lange syndrome . [6] Symptoms [ edit ] The most notable features of Wilson-Turner syndrome are intellectual disability, obesity, hypogonadism, gynecomastia, and distinct facial features. ... The families that were studied and diagnosed with Wilson-Turner Syndrome have shown X-linked recessive pedigree pattern. ... It is studying the possible disturbance of the hypothalamic-pituitary-gonadal axis because of the low levels of androgen are combined with normal levels of FSH and LH . [7] See also [ edit ] Prader–Willi syndrome Fragile X syndrome Börjeson-Forssman-Lehmann syndrome Bardet–Biedl syndrome References [ edit ] ^ a b c d e Harakalova, Magdalena; Boogaard, Marie-Jose van den; Sinke, Richard; Lieshout, Stef van; Tuil, Marc C. van; Duran, Karen; Renkens, Ivo; Terhal, Paulien A.; Kovel, Carolien de (2012-08-01). ... Atlas of X-Linked Intellectual Disability Syndromes . OUP USA. ISBN 9780199811793 . ^ a b "Börjeson-Forssman-Lehman Syndrome - NORD (National Organization for Rare Disorders)" . ... Retrieved 2015-11-01 . ^ "Wilson-Turner X-linked mental retardation syndrome — CheckOrphan" . www.checkorphan.org .
A number sign (#) is used with this entry because of evidence that Wilson-turner X-linked mental retardation syndrome (WTS) is caused by hemizygous mutation in the LAS1L gene (300964) on chromosome Xq12. Description Wilson-Turner syndrome is an X-linked recessive neurologic disorder characterized by intellectual disability, dysmorphic facial features, hypogonadism, short stature, and truncal obesity. ... Some of the features resembled those of Borjeson-Forssman-Lehmann syndrome (BFLS; 301900), but the patients of Wilson et al. (1991) did not have hypermetropia or cataracts in later life and did not have elongated earlobes. ... Mapping By linkage analysis of a family with syndromic X-linked mental retardation, Wilson et al. (1991) found linkage to chromosome Xp21.1-q22 between DXS84 and DXS94. ... The mapping information made the distinction from Borjeson-Forssman-Lehmann syndrome quite definite, inasmuch as the gene for BFLS had been provisionally localized to Xq26-q27.
Wilson-Turner syndrome (WTS) is a very rare X-linked multisystem genetic disease characterized by intellectual disability, truncal obesity, gynecomastia, hypogonadism, dysmorphic facial features, and short stature. Epidemiology Prevalence of WTS is not known. The syndrome has been described in two families to date: 14 males in the 3 most recent generations of the first family, and 7 males and 7 females in a 5-generation Dutch family. ... The described phenotype overlaps with Börjeson-Forssman-Lehmann syndrome, a form of X-linked intellectual disability. Differences between the two described families are small, but there is a possibility that they represent different clinical entities. Etiology The syndrome has been linked to a mutation in the consensus donor splice site of the histonedeacetylase 8 HDAC8 gene (Xq13).
Radioulnar synostosis-microcephaly-scoliosis syndrome, also known as Guiffré-Tsukahara syndrome, is an extremely rare syndrome characterized by the association of radioulnar synostosis with microcephaly, scoliosis, short stature and intellectual deficit.
Gaspar et al. (2008) reported a mother and son with apparent X-linked semidominant inheritance of Giuffre-Tsukahara syndrome. The son was referred at age 7 years because of developmental delay, learning disability, and attention deficit and hyperactivity disorder. ... The authors concluded that Giuffre-Tsukahara syndrome is a single genetic entity characterized by the association of microcephaly and radioulnar synostosis and mental retardation, but without characteristic facies. Dalal et al. (2010) reported a girl, born of consanguineous Indian parents, with apparent Giuffre-Tsukahara syndrome. She presented at age 6 years with global developmental delay, poor speech, microcephaly, short stature, and limited elbow movement. ... Dalal et al. (2010) postulated X-linked dominant inheritance, but noted that autosomal recessive inheritance could not be excluded. Nomenclature The syndrome described here was designated 'Tsukahara syndrome' by Udler et al. (1998). This syndrome is distinct from the 'Tsukahara syndrome' involving type A1 brachydactyly, short stature, scoliosis, microcephaly, ptosis, hearing loss, and mental retardation (613627).
Tsukuhara syndrome Other names Radioulnar synostosis-microcephaly-scoliosis syndrome Specialty Orthopedic Tsukuhara syndrome is an infrequently occurring skeletal dysplasia characterised by a caudal synostosis of the vertebra at birth. [1] References [ edit ] ^ "Radioulnar synostosis with microcephaly, short stature, scoliosis, and mental retardation" .
Smith Martin Dodd syndrome Specialty Cardiology Smith Martin Dodd syndrome is a very rare genetic disorder first described by Smith et al. in 1994. [1] It is characterized by small eyes, a diaphragmatic hernia , and Tetralogy of Fallot , a congenital heart defect . [2] [3] The only known case is of a 9-year-old boy with several congenital anomalies including a diaphragmatic hernia, microphthalmia , and Tetralogy of Fallot. It was found that the boy had a reciprocal translocation t(1;15)(q41;q21.2). [4] A congenital diaphragmatic hernia is consistent with chromosome 1q41-q42 deletion syndrome, [5] and the report by Smith et al. suggested that genes involved in the translocation may be important for the development of morphological characteristics, especially those of the eye or heart. [1] References [ edit ] ^ a b Smith SA, Martin KE, Dodd KL, Young ID (October 1994). ... PMID 7894732 . ^ "Smith Martin Dodd Syndrome" . Check Orphan. Archived from the original on 2012-07-22 . ... Retrieved 2011-10-31 . ^ "FRYNS SYNDROME; FRNS" . Online Medical Inheritance in Man . Retrieved 2011-10-31 . ^ "CHROMOSOME 1q41-q42 DELETION SYNDROME" . Online Medical Inheritance in Man .
Pork–cat syndrome Specialty Immunology Pork–cat syndrome is an allergy to pork , usually after adolescence , that is caused by exposure to cats . Although first described in 1994, [1] [2] [3] it was first documented in the U.S. by Scott Commins and Thomas Platts-Mills during their research on alpha-gal allergy . [4] It is called "pork–cat syndrome" because "almost all people with the condition are cat owners, and many have multiple cats. Some develop an allergic response to cat serum albumin (protein made by a cat’s liver ) that cross-reacts with albumin in pork when someone consumes it, and can lead to severe or even fatal allergic reactions when pork is consumed." [4] References [ edit ] ^ Abreu, Carmo; Gomes, Raquel; Bartolome Borja, Bial-Arístegui; Falcão, Helena; Cunha, Leonor (30 March 2015). "Pork-cat syndrome?" . Clin Transl Allergy . 5 (Suppl 3): P164. doi : 10.1186/2045-7022-5-S3-P164 . PMC 4412402 . ^ "Pork-Cat Syndrome an Under-Recognized Allergy" . ^ Posthumus, Jonathon; James, Hayley R; Lane, Charles J; Matos, Luis A; Platts-Mills, Thomas A E; Commins, Scott P (24 March 2017). "Initial Description of Pork-Cat Syndrome in the United States" . J Allergy Clin Immunol . 131 (3): 923–925. doi : 10.1016/j.jaci.2012.12.665 .
Etiology The etiology of RD is unknown. RD can be part of a syndrome, such as Kallmann syndrome, Bardet-Biedl syndrome, Beckwith-Wiedemann syndrome, diGeorge syndrome, Fraser syndrome, renal coloboma syndrome, and renal cysts and diabetes syndrome (see these terms). HNF1B (17q12), PAX2 (10q24.3-q25.1) and uroplakins genes, which are associated with some of these syndromes, may have an important role in the pathogenesis of RD.