-
Chronic Exertional Compartment Syndrome
Mayo_clinic
Chronic exertional compartment syndrome Chronic exertional compartment syndrome is a musculoskeletal condition brought on by exercise. ... Risk factors Certain factors increase your risk of developing chronic exertional compartment syndrome, including: Age. Although people of any age can develop chronic exertional compartment syndrome, the condition is most common in male and female athletes under age 30. ... Working out too intensely or too frequently also can raise your risk of chronic exertional compartment syndrome. Complications Chronic exertional compartment syndrome isn't a life-threatening condition and usually doesn't cause lasting damage if you get appropriate treatment. ... Treatment Options to treat chronic exertional compartment syndrome include both nonsurgical and surgical methods. ... For chronic exertional compartment syndrome, questions to ask your doctor include: What's the most likely cause of my symptoms?
-
Crush Syndrome
Wikipedia
Earthquakes are a main cause of crush syndrome injuries. Crush syndrome (also traumatic rhabdomyolysis or Bywaters' syndrome ) is a medical condition characterized by major shock and kidney failure after a crushing injury to skeletal muscle . ... Early untreated crush syndrome death is caused by hyperkalemia and by hypovolemic shock . ... ISBN 9780763742393 . ^ Pallister, Ian (20 May 2016). "Management of Compartment Syndrome and Crush Syndrome". Orthopaedic Trauma in the Austere Environment . pp. 363–368. doi : 10.1007/978-3-319-29122-2_28 . ISBN 978-3-319-29120-8 . ^ "Compartment Syndrome - The 5 Ps" . Ausmed. 17 May 2016 . ... PMID 23908797 . ^ a b c d Smith, Jason (23 October 2002). "Crush Injury and Crush Syndrome" . Ovid . 54 . ^ St John Ambulance UK First Aid Manual, 10th Edition, p. 118 ^ "Crush Syndrome" (PDF) .

-
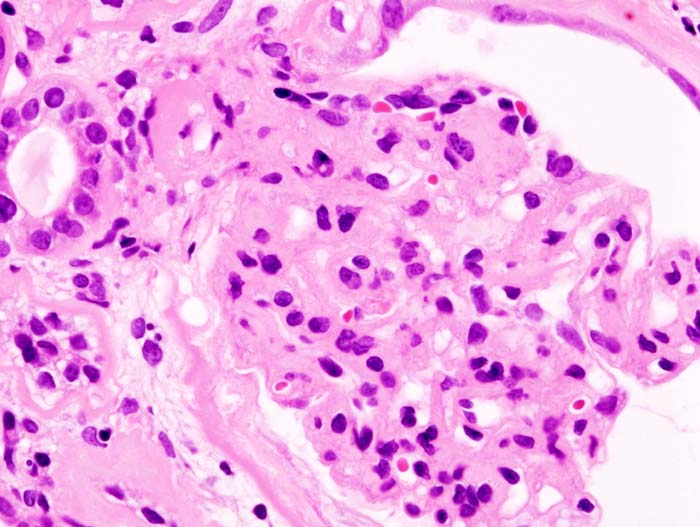
Nephrotic Syndrome
Mayo_clinic
Overview Nephrotic syndrome is a kidney disorder that causes your body to pass too much protein in your urine. ... Nephrotic syndrome can increase your risk of infections and blood clots. ... Chronic kidney disease. Nephrotic syndrome can cause your kidneys to lose their function over time. ... Infections. People with nephrotic syndrome have an increased risk of infections. ... Treatment Treatment for nephrotic syndrome involves treating any medical condition that might be causing your nephrotic syndrome.NPHS2, A2M, SERPINC1, GPC5, HLA-DRB1, ALB, NPHS1, TGFB1, APOA1, ARHGDIA, KANK1, SERPINE1, COL4A1, COL1A1, TNS2, F3, GUCA2B, IL2, LAMA5, HSD11B2, ACE, SOD2, CD2, PODXL, TF, NCK2, FGF2, COL4A2, NCK1, XPO5, NUP37, CTSL, PTGS2, MPV17, IL1B, APOB, ALOX5, ITSN2, FAT1, EDNRA, KCNJ1, ECE1, DDC, GPAM, IL4, NOS1, ACAT1, SCNN1A, MMP1, LIPC, SMAD1, SOAT2, VLDLR, PPARGC1A, DGAT1, CFL1, SCNN1B, PLA2G7, WT1, PLCE1, TRPC6, OSGEP, ITGA3, LAMB2, COQ2, SGPL1, MYO1E, COQ6, MEFV, GATA3, WDR73, SCARB2, NUP107, EMP2, INF2, PTPRO, COL4A5, CRB2, TP53RK, COQ8B, PDSS2, COX2, GSN, CCND1, PAX2, KANK2, ADA, WDR4, DGKE, IL7R, NLRP3, FGA, C3, TBC1D8B, MAGI2, NUP133, ND1, TRNF, TRNH, ND5, ND4, ZFPM2, ND6, PMM2, PTPRD, PARM1, RAG1, COX1, RAG2, MAP3K1, TBX18, RMRP, WWOX, MARS1, ANLN, COX3, TRNS1, TRNL1, IFT27, IFT172, SRY, SLC17A5, SPP1, STAT4, VAMP7, BBS9, TRIM32, SMARCAL1, SERPINA1, SOX9, FOXP3, LAGE3, NPHP1, TPRKB, PRKCD, ZAP70, WDPCP, TRNW, TRNS2, SDCCAG8, LYZ, TRNQ, MKKS, SAA1, MKS1, BBS5, HLA-DQB1, BBS10, CEP290, GLA, GATA4, NR5A1, FN1, ARL6, LMNB2, BBIP1, CHST14, TTC8, LZTFL1, C8orf37, COL4A4, COL4A3, BBS12, CASP10, C1R, C1QA, BTC, BBS4, BBS2, BBS1, B2M, NR0B1, TSBP1-AS1, ACTN4, DCLRE1C, SNAP29, IL2RG, CHD7, BBS7, AHI1, ZNF592, DMRT3, LMX1B, IRAK1, LIG4, LPA, ACTB, CD80, PCSK9, PLG, CD2AP, ABCB1, APOE, NR3C1, APOL1, PLA2R1, TBC1D9, IGAN1, POMC, THSD7A, ANGPTL4, MIF, VEGFA, LEP, MS4A1, AGT, CABIN1, KRT20, IL18, TNF, SOCS3, F9, MIR192, AGTR1, CMIP, CXCL8, IL13, KTWS, HPSE, KIRREL1, BRAF, INS, LPL, MIR30A, REN, RELA, MTHFR, ANGPT2, CFHR5, MIR586, MIR939, MIR146B, AHSA1, HLP, IL21, SQOR, TET1, OLFM1, PPIF, ACE2, DNM1L, CRELD2, SOCS5, IL22, RNF19A, IL23A, LGALSL, PLB1, KANK4, RBM45, GLCCI1, POLDIP2, MLYCD, MUC16, NPNT, ATAD1, NSD3, MIR16-1, APOM, NFASC, MIR23B, HAVCR1, DDN, MZB1, SYNPO, HT, PHB2, TRIM8, FIS1, PIK3CD, GRAP2, ESR1, F2, F5, FKBP5, FLT1, GCG, GLO1, GSTM1, GSTP1, GSTT1, GUCA2A, HDAC2, CFH, HIF1A, HLA-DQA1, HMBS, HP, HPX, HSPA4, HSPB1, IL1A, IL1R1, IL4R, IL6, IL10, IL12B, EXT1, STOM, MAPKAPK2, EDN1, ADCYAP1, APOA4, AQP1, AQP2, AQP4, STS, BCL2, C1S, VPS51, CALR, CD36, CETP, CLU, CNP, CNTN1, COX8A, CLDN7, CRK, MAPK14, CST3, CTSB, CYP2C19, CYP3A4, CYP3A5, HBEGF, IL17A, KDR, KLKB1, LAMP1, RAB5A, RAC1, RYR2, SALL1, CCL2, CCL13, SDC1, SNAI1, STAT3, STAT5A, TFE3, TFPI, TIMP1, TLR4, TP53, TTR, YWHAZ, MANF, AIMP2, FGF23, PDE5A, RAB11A, IQGAP1, CLDN2, CLDN1, PTH, MAPK1, PLCL1, MUC1, LCAT, LDLR, LEPR, LOX, LRP2, LTA4H, MAF, MC1R, MGP, NR3C2, MPO, MYH9, PLAT, MYOC, NOS3, NOTCH1, NPPC, NT5E, PDR, PIK3CA, PIK3CB, PIK3CG, PKD1, PLA2G1B, MTCO2P12
-
Pseudohypoparathyroidism
Wikipedia
This presentation is known as 'knuckle knuckle dimple dimple' sign ( Archibald's sign ). This is as opposed to Turner syndrome which is characterized by blunting of only the fourth knuckle, and Down syndrome , which is associated with a hypoplastic middle phalanx. ... External links [ edit ] Classification D ICD - 10 : E20.1 ICD - 9-CM : 275.49 OMIM : 103580 603233 203330 MeSH : D011547 DiseasesDB : 10835 External resources MedlinePlus : 000364 eMedicine : med/1940 v t e Parathyroid disease Hypoparathyroidism Pseudohypoparathyroidism Pseudopseudohypoparathyroidism Hyperparathyroidism Primary Secondary Tertiary Osteitis fibrosa cystica Other Parathyroiditis v t e Cell surface receptor deficiencies G protein-coupled receptor (including hormone ) Class A TSHR ( Congenital hypothyroidism 1 ) LHCGR ( Luteinizing hormone insensitivity , Leydig cell hypoplasia , Male-limited precocious puberty ) FSHR ( Follicle-stimulating hormone insensitivity , XX gonadal dysgenesis ) GnRHR ( Gonadotropin-releasing hormone insensitivity ) EDNRB ( ABCD syndrome , Waardenburg syndrome 4a , Hirschsprung's disease 2 ) AVPR2 ( Nephrogenic diabetes insipidus 1 ) PTGER2 ( Aspirin-induced asthma ) Class B PTH1R ( Jansen's metaphyseal chondrodysplasia ) Class C CASR ( Familial hypocalciuric hypercalcemia ) Class F FZD4 ( Familial exudative vitreoretinopathy 1 ) Enzyme-linked receptor (including growth factor ) RTK ROR2 ( Robinow syndrome ) FGFR1 ( Pfeiffer syndrome , KAL2 Kallmann syndrome ) FGFR2 ( Apert syndrome , Antley–Bixler syndrome , Pfeiffer syndrome , Crouzon syndrome , Jackson–Weiss syndrome ) FGFR3 ( Achondroplasia , Hypochondroplasia , Thanatophoric dysplasia , Muenke syndrome ) INSR ( Donohue syndrome Rabson–Mendenhall syndrome ) NTRK1 ( Congenital insensitivity to pain with anhidrosis ) KIT ( KIT Piebaldism , Gastrointestinal stromal tumor ) STPK AMHR2 ( Persistent Müllerian duct syndrome II ) TGF beta receptors : Endoglin / Alk-1 / SMAD4 ( Hereditary hemorrhagic telangiectasia ) TGFBR1 / TGFBR2 ( Loeys–Dietz syndrome ) GC GUCY2D ( Leber's congenital amaurosis 1 ) JAK-STAT Type I cytokine receptor : GH ( Laron syndrome ) CSF2RA ( Surfactant metabolism dysfunction 4 ) MPL ( Congenital amegakaryocytic thrombocytopenia ) TNF receptor TNFRSF1A ( TNF receptor associated periodic syndrome ) TNFRSF13B ( Selective immunoglobulin A deficiency 2 ) TNFRSF5 ( Hyper-IgM syndrome type 3 ) TNFRSF13C ( CVID4 ) TNFRSF13B ( CVID2 ) TNFRSF6 ( Autoimmune lymphoproliferative syndrome 1A ) Lipid receptor LRP : LRP2 ( Donnai–Barrow syndrome ) LRP4 ( Cenani–Lenz syndactylism ) LRP5 ( Worth syndrome , Familial exudative vitreoretinopathy 4 , Osteopetrosis 1 ) LDLR ( LDLR Familial hypercholesterolemia ) Other/ungrouped Immunoglobulin superfamily : AGM3, 6 Integrin : LAD1 Glanzmann's thrombasthenia Junctional epidermolysis bullosa with pyloric atresia EDAR ( EDAR hypohidrotic ectodermal dysplasia ) PTCH1 ( Nevoid basal-cell carcinoma syndrome ) BMPR1A ( BMPR1A juvenile polyposis syndrome ) IL2RG ( X-linked severe combined immunodeficiency ) See also cell surface receptors v t e Disorders due to genomic imprinting Chromosome 15 Angelman syndrome ♀ / Prader-Willi syndrome ♂ Chromosome 11 Beckwith–Wiedemann syndrome ♀ / Silver–Russell syndrome ♂ Myoclonic dystonia Chromosome 20 Pseudohypoparathyroidism ♀ / Pseudopseudohypoparathyroidism ♂ Chromosome 6 Transient neonatal diabetes mellitus
-
Reticular Pigmented Anomaly Of The Flexures
Wikipedia
External links [ edit ] Classification D OMIM : 179850 MeSH : C562924 v t e Cytoskeletal defects Microfilaments Myofilament Actin Hypertrophic cardiomyopathy 11 Dilated cardiomyopathy 1AA DFNA20 Nemaline myopathy 3 Myosin Elejalde syndrome Hypertrophic cardiomyopathy 1, 8, 10 Usher syndrome 1B Freeman–Sheldon syndrome DFN A3, 4, 11, 17, 22; B2, 30, 37, 48 May–Hegglin anomaly Troponin Hypertrophic cardiomyopathy 7, 2 Nemaline myopathy 4, 5 Tropomyosin Hypertrophic cardiomyopathy 3 Nemaline myopathy 1 Titin Hypertrophic cardiomyopathy 9 Other Fibrillin Marfan syndrome Weill–Marchesani syndrome Filamin FG syndrome 2 Boomerang dysplasia Larsen syndrome Terminal osseous dysplasia with pigmentary defects IF 1/2 Keratinopathy ( keratosis , keratoderma , hyperkeratosis ): KRT1 Striate palmoplantar keratoderma 3 Epidermolytic hyperkeratosis IHCM KRT2E ( Ichthyosis bullosa of Siemens ) KRT3 ( Meesmann juvenile epithelial corneal dystrophy ) KRT4 ( White sponge nevus ) KRT5 ( Epidermolysis bullosa simplex ) KRT8 ( Familial cirrhosis ) KRT10 ( Epidermolytic hyperkeratosis ) KRT12 ( Meesmann juvenile epithelial corneal dystrophy ) KRT13 ( White sponge nevus ) KRT14 ( Epidermolysis bullosa simplex ) KRT17 ( Steatocystoma multiplex ) KRT18 ( Familial cirrhosis ) KRT81 / KRT83 / KRT86 ( Monilethrix ) Naegeli–Franceschetti–Jadassohn syndrome Reticular pigmented anomaly of the flexures 3 Desmin : Desmin-related myofibrillar myopathy Dilated cardiomyopathy 1I GFAP : Alexander disease Peripherin : Amyotrophic lateral sclerosis 4 Neurofilament : Parkinson's disease Charcot–Marie–Tooth disease 1F, 2E Amyotrophic lateral sclerosis 5 Laminopathy : LMNA Mandibuloacral dysplasia Dunnigan Familial partial lipodystrophy Emery–Dreifuss muscular dystrophy 2 Limb-girdle muscular dystrophy 1B Charcot–Marie–Tooth disease 2B1 LMNB Barraquer–Simons syndrome LEMD3 Buschke–Ollendorff syndrome Osteopoikilosis LBR Pelger–Huet anomaly Hydrops-ectopic calcification-moth-eaten skeletal dysplasia Microtubules Kinesin Charcot–Marie–Tooth disease 2A Hereditary spastic paraplegia 10 Dynein Primary ciliary dyskinesia Short rib-polydactyly syndrome 3 Asphyxiating thoracic dysplasia 3 Other Tauopathy Cavernous venous malformation Membrane Spectrin : Spinocerebellar ataxia 5 Hereditary spherocytosis 2, 3 Hereditary elliptocytosis 2, 3 Ankyrin : Long QT syndrome 4 Hereditary spherocytosis 1 Catenin APC Gardner's syndrome Familial adenomatous polyposis plakoglobin ( Naxos syndrome ) GAN ( Giant axonal neuropathy ) Other desmoplakin : Striate palmoplantar keratoderma 2 Carvajal syndrome Arrhythmogenic right ventricular dysplasia 8 plectin : Epidermolysis bullosa simplex with muscular dystrophy Epidermolysis bullosa simplex of Ogna plakophilin : Skin fragility syndrome Arrhythmogenic right ventricular dysplasia 9 centrosome : PCNT ( Microcephalic osteodysplastic primordial dwarfism type II ) Related topics: Cytoskeletal proteins This cutaneous condition article is a stub .KRT5, POFUT1, POGLUT1, PSENEN, ADAM10, CFH, IL1B, TYRP1, ACAN, CFHR5, TNF, TRP-AGG2-6, RANBP2, DCT, TBPL1, TRPV1, SHBG, LZTR1, TRPC6, SIRT1, TRPC3, TRP-TGG3-1, SMC1A, SYNGAP1, TGFBR2, TGFB1, TAC1, SPTBN2, FTO, OPA1, SET, EGF, ANG, SERPINA6, COL9A2, COL9A3, COMT, CSF1, CCN2, CTNNB1, DDT, EGR3, PTH1R, HNRNPA1, IGF1, IL4, IL5, IL6, CXCL8, LEP, LTA, NFKB1, DDTL
-
Graham-Little Syndrome
Wikipedia
Graham-Little syndrome Specialty Dermatology Graham-Little syndrome is a cutaneous condition characterized by lichen planus -like skin lesions . [1] : 648 It is named after Ernest Graham-Little . [2] See also [ edit ] List of cutaneous conditions References [ edit ] ^ Freedberg, et al. (2003). ... You can help Wikipedia by expanding it . v t e v t e Disorders of skin appendages Nail thickness: Onychogryphosis Onychauxis color: Beau's lines Yellow nail syndrome Leukonychia Azure lunula shape: Koilonychia Nail clubbing behavior: Onychotillomania Onychophagia other: Ingrown nail Anonychia ungrouped: Paronychia Acute Chronic Chevron nail Congenital onychodysplasia of the index fingers Green nails Half and half nails Hangnail Hapalonychia Hook nail Ingrown nail Lichen planus of the nails Longitudinal erythronychia Malalignment of the nail plate Median nail dystrophy Mees' lines Melanonychia Muehrcke's lines Nail–patella syndrome Onychoatrophy Onycholysis Onychomadesis Onychomatricoma Onychomycosis Onychophosis Onychoptosis defluvium Onychorrhexis Onychoschizia Platonychia Pincer nails Plummer's nail Psoriatic nails Pterygium inversum unguis Pterygium unguis Purpura of the nail bed Racquet nail Red lunulae Shell nail syndrome Splinter hemorrhage Spotted lunulae Staining of the nail plate Stippled nails Subungual hematoma Terry's nails Twenty-nail dystrophy Hair Hair loss / Baldness noncicatricial alopecia : Alopecia areata totalis universalis Ophiasis Androgenic alopecia (male-pattern baldness) Hypotrichosis Telogen effluvium Traction alopecia Lichen planopilaris Trichorrhexis nodosa Alopecia neoplastica Anagen effluvium Alopecia mucinosa cicatricial alopecia : Pseudopelade of Brocq Central centrifugal cicatricial alopecia Pressure alopecia Traumatic alopecia Tumor alopecia Hot comb alopecia Perifolliculitis capitis abscedens et suffodiens Graham-Little syndrome Folliculitis decalvans ungrouped: Triangular alopecia Frontal fibrosing alopecia Marie Unna hereditary hypotrichosis Hypertrichosis Hirsutism Acquired localised generalised patterned Congenital generalised localised X-linked Prepubertal Acneiform eruption Acne Acne vulgaris Acne conglobata Acne miliaris necrotica Tropical acne Infantile acne / Neonatal acne Excoriated acne Acne fulminans Acne medicamentosa (e.g., steroid acne ) Halogen acne Iododerma Bromoderma Chloracne Oil acne Tar acne Acne cosmetica Occupational acne Acne aestivalis Acne keloidalis nuchae Acne mechanica Acne with facial edema Pomade acne Acne necrotica Blackhead Lupus miliaris disseminatus faciei Rosacea Perioral dermatitis Granulomatous perioral dermatitis Phymatous rosacea Rhinophyma Blepharophyma Gnathophyma Metophyma Otophyma Papulopustular rosacea Lupoid rosacea Erythrotelangiectatic rosacea Glandular rosacea Gram-negative rosacea Steroid rosacea Ocular rosacea Persistent edema of rosacea Rosacea conglobata variants Periorificial dermatitis Pyoderma faciale Ungrouped Granulomatous facial dermatitis Idiopathic facial aseptic granuloma Periorbital dermatitis SAPHO syndrome Follicular cysts " Sebaceous cyst " Epidermoid cyst Trichilemmal cyst Steatocystoma simplex multiplex Milia Inflammation Folliculitis Folliculitis nares perforans Tufted folliculitis Pseudofolliculitis barbae Hidradenitis Hidradenitis suppurativa Recurrent palmoplantar hidradenitis Neutrophilic eccrine hidradenitis Ungrouped Acrokeratosis paraneoplastica of Bazex Acroosteolysis Bubble hair deformity Disseminate and recurrent infundibulofolliculitis Erosive pustular dermatitis of the scalp Erythromelanosis follicularis faciei et colli Hair casts Hair follicle nevus Intermittent hair–follicle dystrophy Keratosis pilaris atropicans Kinking hair Koenen's tumor Lichen planopilaris Lichen spinulosus Loose anagen syndrome Menkes kinky hair syndrome Monilethrix Parakeratosis pustulosa Pili ( Pili annulati Pili bifurcati Pili multigemini Pili pseudoannulati Pili torti ) Pityriasis amiantacea Plica neuropathica Poliosis Rubinstein–Taybi syndrome Setleis syndrome Traumatic anserine folliculosis Trichomegaly Trichomycosis axillaris Trichorrhexis ( Trichorrhexis invaginata Trichorrhexis nodosa ) Trichostasis spinulosa Uncombable hair syndrome Wooly hair nevus Sweat glands Eccrine Miliaria Colloid milium Miliaria crystalline Miliaria profunda Miliaria pustulosa Miliaria rubra Occlusion miliaria Postmiliarial hypohidrosis Granulosis rubra nasi Ross’ syndrome Anhidrosis Hyperhidrosis Generalized Gustatory Palmoplantar Apocrine Body odor Chromhidrosis Fox–Fordyce disease Sebaceous Sebaceous hyperplasia
-
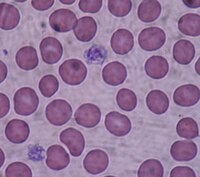
Giant Platelet Disorder
Wikipedia
Giant platelet disorder occurs for inherited diseases like Bernard–Soulier syndrome , gray platelet syndrome and May–Hegglin anomaly . [1] Contents 1 Signs and symptoms 2 Genetics 3 Diagnosis 3.1 Classification 4 Treatment 5 References 6 External links Signs and symptoms [ edit ] Symptoms usually present from the period of birth to early childhood as: nose bleeds, bruising, and/or gum bleeding. Problems later in life may arise from anything that can cause internal bleeding such as: stomach ulcers, surgery, trauma, or menstruation. [2] Abnormality of the abdomen, nosebleeds , heavy menstrual bleeding , purpura , too few platelets circulating in the blood , and prolonged bleeding time have also been listed as symptoms of various giant platelet disorders. [3] Genetics [ edit ] Many of the further classifications of giant platelet disorder occur as a result of being genetically passed down through families as an autosomal recessive disorder, such as in Bernard-Soulier syndrome and gray platelet syndrome. [4] Diagnosis [ edit ] People may be diagnosed after prolonged and/or recurring bleeding episodes. ... This would utilize platelet aggregation studies and flow cytometry. [5] Classification [ edit ] Giant platelet disorders can be further categorized: [6] caused by auto-immune disorders, for example Immune thrombocytopenic purpura (ITP), and characterized by low platelet count, but high MPV (mean platelet volume). [7] Caused by glycoprotein abnormalities: Bernard–Soulier syndrome , velocardiofacial syndrome Caused by calpain defect: Montreal platelet syndrome Caused by alpha granules defect: gray platelet syndrome Characterized by abnormal neutrophil inclusions: May–Hegglin anomaly , Sebastian syndrome With systemic manifestations: Hereditary macrothrombocytopenia with hearing loss, Epstein syndrome , Fechtner syndrome With no specific abnormalities: Mediterranean macrothrombocytopenia Harris platelet syndrome Treatment [ edit ] There has been no general recommendation for treatment of patients with giant platelet disorders, as there are many different specific classifications to further categorize this disorder which each need differing treatments. ... Retrieved 13 November 2017 . ^ "Giant platelet syndrome" . MedicineNet . Retrieved 2016-04-14 . ^ "Giant platelet syndrome | Disease | Symptoms | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . ... PMID 20011639 . ^ "Bernard–Soulier Disease (Giant Platelet Syndrome) Symptoms, Causes, Treatment - What is Bernard–Soulier syndrome?
-
Carpenter Syndrome
Medlineplus
Carpenter syndrome is a condition characterized by the premature fusion of certain skull bones (craniosynostosis), abnormalities of the fingers and toes, and other developmental problems. ... People with Carpenter syndrome often have intellectual disability, which can range from mild to profound. ... The life expectancy for individuals with Carpenter syndrome is shortened but extremely variable. The signs and symptoms of Carpenter syndrome are similar to another genetic condition called Greig cephalopolysyndactyly syndrome. ... It is unclear how disruptions in protein function lead to the features of Carpenter syndrome, but it is likely that interference with normal body patterning plays a role.
-
Morquio Syndrome C
Omim
There is some evidence of an additional form of Morquio syndrome, referred to here as type C, in which urinary excretion of keratan sulfate is absent. However, McKusick (1972) suggested that the nonkeratosulfate- excreting Morquio syndrome may be allelic to other forms of Morquio syndrome. ... These patients belonged to a consanguineous kindred of Early Americans in southern Maryland in which several autosomal recessive disorders, including Crigler-Najjar syndrome (218800), had been observed. McKusick (1972) suggested that the condition in this family represented a 'nonkeratansulfate-excreting' form of Morquio syndrome, which he postulated was likely allelic to other forms of Morquio syndrome. ... Maroteaux et al. (1982) described 2 children, aged 8 and 7 years, who had a mild form of Morquio syndrome and absence of urinary keratosulfate excretion. ... Beck et al. (1986) suggested that this disorder may represent a 'type C' Morquio syndrome. Fujimoto and Horwitz (1983) studied 2 offspring of second-cousin Mexican parents described as having 'nonkeratosulfate-excreting Morquio syndrome.'
-
Fanconi Renotubular Syndrome 1
Omim
Description Fanconi renotubular syndrome is a consequence of decreased solute and water reabsorption in the proximal tubule of the kidney. ... Genetic Heterogeneity of Fanconi Renotubular Syndrome Fanconi renotubular syndrome-1 has been mapped to chromosome 15q15.3. ... Smith et al. (1976) described a kindred in which the syndrome appeared in 4 successive generations and was possibly associated with diabetes mellitus. ... Tolaymat et al. (1992) stated that 10 families with Fanconi syndrome had been described, of which 6 had an autosomal dominant mode of transmission. ... Inheritance The transmission pattern of renal Fanconi syndrome in the family reported by Wen et al. (1989) was consistent with autosomal dominant inheritance.
-
Long Qt Syndrome 5
Omim
A number sign (#) is used with this entry because of evidence that long QT syndrome-5 (LQT5) is caused by heterozygous mutation in the KCNE1 gene (176261) on chromosome 21q22. ... For a discussion of genetic heterogeneity of long QT syndrome, see LQT1 (192500). Molecular Genetics In affected members of 2 families with long QT syndrome, Splawski et al. (1997) identified heterozygosity for different missense mutations in the KCNE1 gene (176261.0003-176261.0004). Splawski et al. (2000) screened 262 unrelated individuals with LQT syndrome for mutations in the 5 defined genes (KCNQ1; KCNH2; SCN5A, 600163; KCNE1; and KCNE2 603796) and identified mutations in 177 individuals (68%). KCNQ1 and KCNH2 accounted for 87% of mutations (42% and 45%, respectively), and SCN5A, KCNE1, and KCNE2 for the remaining 13% (8%, 3%, and 2%, respectively). Acquired Long QT Syndrome Paulussen et al. (2004) screened 5 congenital long QT syndrome-associated genes (KCNQ1, KCNH2, SCN5A, KNCE1, and KCNE2) in 32 individuals with drug-induced long QT syndrome and identified 3 heterozygous mutations in 4 patients that were not found in 32 healthy controls (see, e.g., 176261.0005). ... In 44 unrelated patients with LQT syndrome, Millat et al. (2006) used DHLP chromatography to analyze the KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 genes for mutations and SNPs.
-
Hellp Syndrome
Wikipedia
Class II HELLP syndrome is characterised by a platelet count of 50,000-100,000/µL. Class III HELLP syndrome is characterised by a platelet count of 100,000-150,000/µL. ... Postpartum occurrences are also observed in 30% of all HELLP syndrome cases. [51] History [ edit ] HELLP syndrome was identified as a distinct clinical entity (as opposed to severe pre-eclampsia) by Dr. ... "DCurrent understanding of severe preeclampsia, pregnancy-associated hemolytic uremic syndrome, thrombotic thrombocytopenic purpura, hemolysis, elevated liver enzymes, and low platelet syndrome, and postpartum acute renal failure: different clinical syndromes or just different names?". ... "Hepatic hemorrhage and the HELLP syndrome: a surgeon's perspective". Am Surg . 61 (9): 756–60.CD46, CFH, CFI, HELLPAR, FASLG, FAS, PGF, F5, LEP, HADHA, F2, TNF, HPGDS, LGALS13, FLT1, VEGFA, MTHFR, MAPK14, TLR4, AIMP2, TLR2, MAPK3, TPBG, VEGFC, TGFB3, VWF, MAPK1, ABCG2, TFPI2, IL18R1, GRAP2, EBI3, AHSA1, ADAMTS13, SIRT4, RNF19A, POLDIP2, SLC17A5, ERVW-1, MBL3P, AHSP, NOD2, POTEF, SERPINE2, ACTB, SERPINE1, PAH, APC, CFB, CA9, CD40LG, CD59, CDKN1C, COX8A, CP, CRK, ENG, EPHX1, GAPDH, GNB3, GPT, NR3C1, GSTM1, GSTT1, HSPA4, HSPG2, IFNG, IL1B, IL1RN, CXCL8, IL10, LEPR, LNPEP, ADM, NOS3, PAEP, MBL2
-
Anterior Cerebral Artery Syndrome
Wikipedia
Anterior cerebral artery syndrome Outer surface of cerebral hemisphere, showing areas supplied by cerebral arteries. ... Specialty Neurology Anterior cerebral artery syndrome is a condition whereby the blood supply from the anterior cerebral artery (ACA) is restricted, leading to a reduction of the function of the portions of the brain supplied by that vessel: the medial aspects of the frontal and parietal lobes, basal ganglia , anterior fornix and anterior corpus callosum . [1] Depending upon the area and severity of the occlusion, signs and symptoms may vary within the population affected with ACA syndrome. ... Homolateral ataxia and crural paresis: a syndrome of anterior cerebral artery territory infarction. ... Atypical presentations of acute cerebrovascular syndromes. Lancet Neurol. 2011 Jun;10(6):550-60. ... ISBN 0-8036-1247-8 External links [ edit ] Classification D ICD - 10 : G46.1 MeSH : D020243 v t e Cerebrovascular diseases including stroke Ischaemic stroke Brain Anterior cerebral artery syndrome Middle cerebral artery syndrome Posterior cerebral artery syndrome Amaurosis fugax Moyamoya disease Dejerine–Roussy syndrome Watershed stroke Lacunar stroke Brain stem Brainstem stroke syndrome Medulla Medial medullary syndrome Lateral medullary syndrome Pons Medial pontine syndrome / Foville's Lateral pontine syndrome / Millard-Gubler Midbrain Weber's syndrome Benedikt syndrome Claude's syndrome Cerebellum Cerebellar stroke syndrome Extracranial arteries Carotid artery stenosis precerebral Anterior spinal artery syndrome Vertebrobasilar insufficiency Subclavian steal syndrome Classification Brain ischemia Cerebral infarction Classification Transient ischemic attack Total anterior circulation infarct Partial anterior circulation infarct Other CADASIL Binswanger's disease Transient global amnesia Haemorrhagic stroke Extra-axial Epidural Subdural Subarachnoid Cerebral/Intra-axial Intraventricular Brainstem Duret haemorrhages General Intracranial hemorrhage Aneurysm Intracranial aneurysm Charcot–Bouchard aneurysm Other Cerebral vasculitis Cerebral venous sinus thrombosis v t e Symptoms , signs and syndromes associated with lesions of the brain and brainstem Brainstem Medulla (CN 8, 9, 10, 12) Lateral medullary syndrome/Wallenberg PICA Medial medullary syndrome/Dejerine ASA Pons (CN 5, 6, 7, 8) Upper dorsal pontine syndrome/Raymond-Céstan syndrome Lateral pontine syndrome ( AICA ) (lateral) Medial pontine syndrome / Millard–Gubler syndrome / Foville's syndrome ( basilar ) Locked-in syndrome Internuclear ophthalmoplegia One and a half syndrome Midbrain (CN 3, 4) Weber's syndrome ventral peduncle, PCA Benedikt syndrome ventral tegmentum, PCA Parinaud's syndrome dorsal, tumor Claude's syndrome Other Alternating hemiplegia Cerebellum Latearl Dysmetria Dysdiadochokinesia Intention tremor ) Medial Cerebellar ataxia Basal ganglia Chorea Dystonia Parkinson's disease Cortex ACA syndrome MCA syndrome PCA syndrome Frontal lobe Expressive aphasia Abulia Parietal lobe Receptive aphasia Hemispatial neglect Gerstmann syndrome Astereognosis Occipital lobe Bálint's syndrome Cortical blindness Pure alexia Temporal lobe Cortical deafness Prosopagnosia Thalamus Thalamic syndrome Other Upper motor neuron lesion Aphasia

-
Hypotonia
Wikipedia
Hypotonia Other names Floppy baby syndrome An infant with botulism ; despite not being asleep or sedated, he cannot open his eyes or move; he also has a weak cry. ... For instance, some people with hypotonia may experience constipation, while others have no bowel problems. [ citation needed ] Floppy baby syndrome [ edit ] The term "floppy infant syndrome" is used to describe abnormal limpness when an infant is born. ... Because these low-toned muscles do not fully contract before they again relax (muscle accommodates to the stimulus and so shuts down again), they remain loose and very stretchy, never realizing their full potential of maintaining a muscle contraction over time. " Cause [ edit ] Some conditions known to cause hypotonia include: Congenital – i.e. disease a person is born with (including genetic disorders presenting within 6 months) Genetic disorders are the most common cause 22q13 deletion syndrome a.k.a. Phelan–McDermid syndrome 3-Methylcrotonyl-CoA carboxylase deficiency [4] Achondroplasia Aicardi syndrome Autism spectrum disorders [5] Canavan disease Centronuclear myopathy (including myotubular myopathy) Central core disease CHARGE syndrome Cohen syndrome Costello syndrome Dejerine–Sottas disease (HMSN Type III) Down syndrome a.k.a. trisomy 21 — most common Ehlers–Danlos syndrome Familial dysautonomia (Riley–Day syndrome) FG syndrome Fragile X syndrome Griscelli syndrome Type 1 (Elejalde syndrome) Disorder Growth Hormone Disorder Pituitary Dwarfism Holocarboxylase synthetase deficiency / Multiple carboxylase deficiency [6] Krabbe disease Leigh's disease Lesch–Nyhan syndrome [7] Marfan's syndrome Menkes syndrome Methylmalonic acidemia Myotonic dystrophy Niemann–Pick disease Nonketotic hyperglycinemia (NKH) or Glycine encephalopathy (GCE) Noonan syndrome Neurofibromatosis Patau syndrome a.k.a. trisomy 13 Prader–Willi syndrome Rett syndrome Septo-optic dysplasia (de Morsier syndrome) Snyder–Robinson syndrome (SRS) Spinal muscular atrophy (SMA) Succinic semialdehyde dehydrogenase deficiency (SSADH) Tay–Sachs disease Werdnig–Hoffmann syndrome – Spinal muscular atrophy with congenital degeneration of anterior horns of spinal cord. Autosomal recessive [8] Wiedemann–Steiner syndrome Williams syndrome Zellweger syndrome a.k.a. cerebrohepatorenal syndrome Developmental disability Cerebellar ataxia (congenital) Sensory processing disorder Developmental coordination disorder Hypothyroidism (congenital) Hypotonic cerebral palsy Teratogenesis from in utero exposure to Benzodiazepines Acquired [ edit ] Acquired – i.e. onset occurs after birth Genetic Muscular dystrophy (including Myotonic dystrophy ) – most common Metachromatic leukodystrophy Rett syndrome Spinal muscular atrophy Infections Encephalitis Guillain–Barré syndrome Infant botulism Meningitis Poliomyelitis Sepsis Toxins Infantile acrodynia (childhood mercury poisoning ) Autoimmunity disorders Myasthenia gravis – most common Abnormal vaccine reaction Celiac disease [9] Metabolic disorder Hypervitaminosis Kernicterus Rickets Neurological Traumatic brain injury , such as the damage that is caused by shaken baby syndrome Lower motor neuron lesions Upper motor neuron lesions Miscellaneous Central nervous system dysfunction, including cerebellar lesions and cerebral palsy Hypothyroidism Sandifer syndrome Neonatal benzodiazepine withdrawal syndrome in children born to mothers treated in late pregnancy with benzodiazepine medications [10] Diagnosis [ edit ] The approach to diagnosing the cause of hypotonia (as with all syndromes in neurology) is first localization. ... Other terms for the condition include: [ citation needed ] Low Muscle Tone Benign Congenital Hypotonia Congenital Hypotonia Congenital Muscle Hypotonia Congenital Muscle Weakness Amyotonia Congenita Floppy Baby Syndrome Infantile Hypotonia Prognosis and treatment [ edit ] This section needs more medical references for verification or relies too heavily on primary sources .NSD2, MSL3, RRM2B, MECP2, LETM1, KANSL1, NUDT2, PPP2CA, PAX7, INTS1, ASCC1, LSS, P4HTM, DDC, CHST14, RALGAPA1, ABAT, SLC25A3, MPZ, CDKL5, PREPL, GLDC, NDUFS7, AP4B1-AS1, SYNE2, VPS13A, KIAA0556, CAMTA1, SATB2, RPGRIP1L, CLASP1, SYNE1, SPECC1L, ADNP, MED13L, KANK1, MLYCD, CRB1, ETHE1, KAT6B, RBFOX2, ATP6V0A2, PIGN, LEMD3, KCNE5, AIPL1, FLRT3, PMPCA, CIC, CIZ1, PHF8, RAI1, ZMYND11, WDR4, SRCAP, PNPLA6, MAGED2, TTN-AS1, TLK2, B4GAT1, WDR45, SRD5A3-AS1, AP4S1, PLPBP, B4GALT7, MRAS, FASTKD2, FAN1, RAB3GAP1, RAB18, CLUAP1, EMC1, ZNF423, IQSEC2, POGZ, PLXND1, RAB3GAP2, NDUFAF3, NIPBL, KMT5B, ALG6, POMT2, PET100, DUOX2, TPRKB, CRPPA, EXOSC3, YARS2, NDUFA13, MLXIPL, SEPSECS, NDUFAF1, ZDHHC9, CTCF, TACO1, TMEM216, TIMMDC1, KCNK9, WAC, MBTPS2, TMEM138, NBAS, LIPT1, KLF13, PTRH2, ATP8A2, GMPPA, GMPPB, ANKRD11, NDUFAF4, SIN3A, SH2B1, PARS2, MMACHC, NSMF, SETBP1, KIFBP, RNU4ATAC, B3GAT3, FBXL4, HIBCH, VPS33B, SACS, SLC17A5, TBL2, RPS6KC1, MLH3, ACAD8, B9D1, BBS9, AHDC1, MMADHC, CCDC22, FLVCR1, SETD2, AP4B1, POMT1, MAP3K20, HESX1, OFD1, PPM1D, PEX3, CNTNAP1, KCNAB2, IKBKG, ELP1, CDK13, PEX11B, SUCLG1, SUCLA2, DPM1, FGF17, NPC2, SYNGAP1, ST3GAL5, EIF2B4, EIF2B3, EIF2B2, EIF2B5, AP1S2, PHOX2B, BAZ1B, SEMA5A, SLC7A7, PIGQ, CUL3, CUL4B, GNPAT, PLA2G6, NELFA, YWHAE, ZIC1, PCGF2, BSND, MOGS, TUBA1A, PAX8, BRPF1, ALDH5A1, KAT6A, LHX3, SHOC2, KMT2D, AAAS, HMGA2, DGCR6, LZTR1, ESS2, USP9X, RBM10, SMC1A, LAGE3, ARID1A, COLQ, TBX19, SMC3, BUB3, KIAA0586, ZEB2, SEC24D, FIG4, WASHC5, AMMECR1, MED12, DGCR2, SCO2, AKT3, FARSB, LRPPRC, AASS, ALG3, CD96, COQ7, HNRNPR, APC2, LAMC3, RXYLT1, SEMA3A, TUBB4A, PIBF1, COG5, ZBTB18, DEAF1, PTDSS1, TMEM94, HACD1, HDAC4, LARGE1, LRAT, ADGRG1, TRIP13, TRIP12, SNAP29, LONP1, COG1, HS6ST1, PEX16, EIF2AK3, CHST3, PIGL, TBX4, ADAMTS2, MPDU1, GTF2IRD1, SEC24C, ZNF592, IQCB1, CEP57, RUBCN, SEMA3E, CEP104, IFT140, INPP5K, BCL11A, HCN1, CEP41, RFT1, NDUFAF2, COG7, MTSS2, GDF6, STRADA, GORAB, TIMM50, PGAP3, TRMT10A, TBCK, FOXP2, PRRT2, EARS2, STX1B, FDX2, TP53RK, NACC1, MYMK, SLC46A1, SLC52A3, TOE1, CCDC151, COX20, ANTXR2, CYP2R1, ATPAF2, PKDCC, TMEM67, UBE3B, PUS1, WNT10A, SLC19A3, ASXL3, SLC2A10, CLPB, TRIM8, SPRY4, LAS1L, TMEM47, TRAPPC9, ADGRV1, POMK, PHF6, COA8, COG8, KISS1R, KDM2B, ORAI1, POMGNT2, WDR73, COX14, USP45, ATCAY, LHX4, MTFMT, METTL23, NUBPL, IYD, TUBB, HYLS1, HEPACAM, JMJD1C, STAC3, EBF3, FEZF1, BRWD3, NALCN, ASPM, MYO1H, CHAMP1, SLC13A5, CANT1, LAMA1, WDR62, CCDC141, DOK7, MMAB, FLG-AS1, SLC6A19, COA6, NEXMIF, RD3, ZFP57, TUBB2B, ARL13B, UBR1, ARID2, ASXL1, SPNS2, WDR81, NDUFA11, B3GALT6, DMBX1, TBC1D20, PROKR2, DIS3L2, NDUFAF6, AMER1, PTCHD1, SLC34A3, A2ML1, RDH12, B3GALNT2, SIK1, CKAP2L, CEP120, VPS13B, FAM120AOS, SPRED1, LGI4, MMAA, LCA5, ARX, WDR26, ARMC9, NANS, ALG1, PACS1, WDR11, NUP133, AGK, NGLY1, MBD5, LMBRD1, SPATA7, TMEM126B, HDAC8, ZC4H2, NDUFA12, INPP5E, PRR12, TWNK, MCCC1, MRPS22, COQ8A, HYMAI, RPGRIP1, C12orf4, NUP107, THOC2, SELENON, MCOLN1, THAP11, PEX26, PIGV, OSGEP, CHD7, RIN2, D2HGDH, RETREG1, RNF216, DGCR8, BDNF-AS, TMCO1, CLIP2, MAGEL2, NR2F1-AS1, DUOXA2, PDP1, IL17RD, AHI1, MKS1, TMEM70, OXSM, RMND1, FKBP14, DARS2, SETD5, ASXL2, SLC35C1, FOXRED1, POMGNT1, TBC1D24, ARID1B, CEP290, TCTN1, TMEM237, UPF3B, WNK1, CPLANE1, ALG12, NDUFAF5, FKRP, GNPTAB, TMEM43, THOC6, SLC52A2, TMEM231, SRD5A3, ARHGAP31, TBL1XR1, BBS10, SLC25A22, ALG9, EHMT1, CAMKMT, CSPP1, TCTN2, ALG13, L2HGDH, CENPT, NAA15, BCL11B, NMNAT1, SIL1, LMBR1, HECW2, PCDH19, HACE1, CC2D2A, ALS2, CHD8, ZSWIM6, PRX, EPG5, WDR19, PRUNE1, KMT2C, TRPV4, SLC25A19, PROK2, TRAPPC11, PIEZO2, ZNF335, PRDM16, MCCC2, IFIH1, IRF2BPL, SEMA4A, STRA6, NSD1, NDUFB11, UFD1, VRK1, GAMT, FLT4, FMR1, MTOR, FUCA1, GAA, GABRA1, GABRB3, GABRD, GABRG2, GALC, GALE, GALNS, GATA1, FLG, GBA, GBE1, GCDH, GCK, GCSH, GDI1, GDNF, B4GALT1, GJA1, GJA8, GJB1, GPC3, FLII, FOXE1, GLI2, ETFB, EIF2B1, EIF2S3, ELN, EMD, EP300, CLN8, ERCC1, ERCC2, ERCC5, ERCC6, ERF, ETFA, ETFDH, FOXG1, EXT1, EZH2, ACSL4, FBN1, FBP1, FKTN, GPC4, FGF8, FGFR1, FGFR3, FH, FHL1, GLE1, GM2A, VLDLR, KCNB1, IDUA, IMPDH1, INS, INPPL1, PDX1, ITGA3, ITGA7, ITGB6, ITPR1, ANOS1, KARS1, KCNA2, KCNC3, HSPD1, KCNH1, KCNJ10, KCNJ11, KCNJ13, KCNQ2, KCNQ3, KIT, KIF11, KIF22, KRAS, LAMA2, LAMB2, IDH2, HRAS, GNAO1, HSD17B10, GNB1, GP1BB, GRID2, GRIN1, GRIN2B, GRM7, MSH6, GTF2I, GUSB, GUCY2D, H1-4, H3-3A, HADHA, HPRT1, HADHB, HARS1, HBA1, HBA2, HBB, HEXA, HLCS, HMGCL, HNRNPH2, HNRNPK, HNRNPU, HPD, EGR2, EFNB1, EEF1A2, BMP2, ATP2B3, ALDH7A1, ATP6V1A, ATP6AP1, ATP7A, ATRX, KIF1A, BBS2, BCKDHA, BCKDHB, BCS1L, BDNF, BMPR1A, RERE, BRAF, BRCA2, BTD, BUB1, BUB1B, CA8, CACNA1A, CACNA1C, CAMK2A, CAMK2B, CASR, CDKN1C, ATP1A3, ATIC, EDNRB, AGA, ACADM, ACADS, ACADSB, ACADVL, ACAT1, ACOX1, ACP2, ACTA1, ACTB, ACY1, ADCY5, ADCY6, AHSG, ASPA, ALAD, ALDH3A2, ALPL, AMT, ANK3, SLC25A4, FAS, AR, ARL3, ARSA, ARVCF, ASCL1, CFL2, CHRNA1, CHRNA7, DNM1, CYP27B1, DAG1, DBH, DBT, DCC, DCX, DDX3X, DES, DGUOK, DHCR7, DLG3, DMD, DMPK, CHRNB2, DNAH5, DYNC1H1, DNMT3A, DOCK3, DPAGT1, DPYD, SLC26A2, DUSP6, DYRK1A, TOR1A, ECHS1, EDN3, CTNND2, CTNNB1, NKX2-5, CSTB, CLCNKA, CLCNKB, CLTC, COL1A1, COL1A2, COL2A1, COL4A1, COL4A2, COL5A1, COL5A2, COL6A2, COL6A3, COL12A1, COMT, COX6B1, COX8A, COX10, COX15, CPS1, CPT1A, CPT2, CRAT, CREBBP, CRX, CRYBA1, LIFR, LIMK1, LMNA, SCN9A, RRAS, RREB1, RYR1, MSMO1, SC5D, ATXN1, ATXN2, SCN1A, SCN1B, SCN2A, SCN4A, SCN8A, SCO1, RPS6KA3, SDHA, SET, SIM1, SIX3, SKI, SLC2A1, SLC3A1, SLC5A5, SLC6A1, SLC6A8, SLC16A2, SLC18A2, RPS20, RPL10, PSAP, RAD21, PTCH1, PTEN, PTPN11, PTS, PURA, PEX19, PEX2, PEX5, PYCR1, ALDH18A1, QDPR, RAC1, RAD51, RPE65, RAF1, RAP1A, RAP1B, RARB, RASA2, RB1, DPF2, RET, REV3L, RFC2, RIT1, RMRP, SLC22A5, SNAI2, SMARCA4, TPO, NR2F1, TG, TGFB2, TGFBR2, THRA, THRB, NKX2-1, TK2, TNXB, TPI1, TPM2, TPM3, TRPS1, SMARCB1, TSHB, TSHR, TTN, TULP1, HIRA, UBA1, UBE2A, UBE3A, ABCB7, SLC35A2, KDM6A, VDR, TCF20, TCF4, TBX1, TACR3, SMARCC2, SMARCE1, SMN1, SMN2, SMPD1, SMS, SON, SOS1, SOS2, SOX2, SOX4, SOX5, SOX9, SOX10, SOX11, SPARC, SPAST, SPG7, SPTBN2, STAT3, STIM1, STXBP1, ABCC8, SURF1, SYT1, LRP5, PSMD12, RELN, NDUFA4, TRNS2, TRNW, TRIM37, MMUT, MVK, MYBPC3, MYL2, MYO5A, NAGA, NDP, NDUFA1, NDUFA2, NDUFA6, TRNQ, NEB, NDUFA9, NDUFA10, NDUFB3, NDUFB9, NDUFB10, NDUFS1, NDUFS2, NDUFS3, NDUFV1, NDUFS4, NDUFS6, TRNS1, TRNN, PRPS1, COX1, LTC4S, EPCAM, MAN2B1, MEF2C, MIPEP, MLH1, KMT2A, MLLT1, MN1, MPI, MSH2, ATP8, COX2, TRNL1, COX3, MTM1, ND1, ND2, ND3, ND4, ND5, ND6, MTTP, MTRR, TRNF, TRNH, NDUFS8, NDUFV2, NEU1, PMP22, PEX10, PEX12, PEX13, PEX14, PHKG2, PHYH, PIK3CA, PLAGL1, PLCB4, PLOD1, PLP1, PMM2, PMS1, NF1, PMS2, POLE, POLG, POU1F1, PPM1B, PPP2R1A, PPP2R5D, PPT1, MAP2K1, MAP2K2, PRODH, PROP1, PEX7, PEX6, PEX1, SLC26A4, NFIX, TONSL, CNOT3, NOTCH1, NOTCH3, PNP, NPC1, NPHP1, NRAS, OCA2, OGDH, OPA1, OPHN1, SIX6, OTX2, P4HB, PAFAH1B1, PRDX1, PAH, PAX6, PC, PCBD1, PCYT1A, PDHA1, EMC1-AS1, ATXN7, ABL1

-
Tethered Spinal Cord Syndrome
Wikipedia
TCS may also be related to Ehlers–Danlos syndrome , or Klippel–Feil syndrome , which should also be screened for upon a positive TCS diagnosis. ... "Ultrasonic Evaluation of Tethered Cord Syndrome". In Yamada, Shokei (ed.). Tethered Cord Syndrome in Children and Adults (Rev. ed.). ... "Clinical Experience in Urological Involvement with Tethered Cord Syndrome". In Yamada, Shokei (ed.). Tethered Cord Syndrome in Children and Adults (Rev. ed.). ... "Lower Urinary Tract Dysfunction in Tethered Cord Syndrome". In Yamada, Shokei (ed.). Tethered Cord Syndrome in Children and Adults (Rev. ed.). ... "Lower Urinary Tract Dysfunction in Tethered Cord Syndrome". In Yamada, Shokei (ed.). Tethered Cord Syndrome in Children and Adults (Rev. ed.).MTHFR, PAX3, PDGFRA, VANGL2, PCMT1, CFL1, PON1, CHKA, CCL2, TXN2, PCYT1A, FGFR2, RNF2, SUZ12, MTRR, MTR, MTHFD1, BRCA1, SLC25A1, PTCH1, DLL4, FLI1, FANCL, RFWD3, FANCI, TRIM36, FANCC, SUFU, PSAT1, UBE2T, PHGDH, FANCF, HAAO, MEOX1, PTCH2, MAD2L2, LMX1B, RBM8A, BMS1, KIF22, FANCG, GDF3, FANCB, CREBBP, BRIP1, BRCA2, RAD51C, SLX4, RAD51, PALB2, FANCE, RMRP, PORCN, XRCC2, GDF6, SLC25A19, FANCM, ERCC4, FANCA, FANCD2, ZIC2, DHFR, GRHL3, FUZ, BHMT, CELSR1, TYMS, PTK7, CBS, NOS3, LEPR, NAT1, PAX1, PRCP, SLC19A1, TIMP2, CYP26A1, GRHL2, CBSL, AFP, PGPEP1, FOLR1, FOLR2, SCRIB, COMT, BMP1, ZIC1, VSIR, PKD2L1, VANGL1, ANKEF1, CD200R1, PRICKLE2, UCP2, PCSK9, TP53, SLC5A11, SLC39A4, CCDC88A, ALDH1A2, ISYNA1, TGIF1, TRIM26, FZD3, FKBP8, BHMT2, GPR161, CUBN, DVL1P1, FOXN1, ALDH1L1, MTHFD2, TNIP1, NKX2-8, NNMT, TGFB1, SOX3, GCKR, FOSB, FOS, FOLH1, FLT4, GPC5, FGF3, FAP, ERCC2, EPHA4, DVL3, DVL1, DPP4, TRDMT1, SARDH, CSF2, CRABP2, ABCC2, CD38, CASP8, CASP3, BMP4, BDNF, ARSA, APOE, APOB, APEX1, APAF1, ABO, GLI2, GNAS, UTS2R, LAMC2, SLC2A1, SKI, RFC1, PRKCB, PRKCA, NAT2, NGF, MS, CD200, MMP2, LRP6, LMNB1, LEP, JUND, HK1, JUNB, JUN, JARID2, ITPK1, IL18, IL13, IL10, HOXD@, HOXC@, HOXB@, HOXA@, HMOX1, HLA-DRB4, RN7SL263P
-
Churg-Strauss Syndrome
Mayo_clinic
Overview Churg-Strauss syndrome is a disorder marked by blood vessel inflammation. ... Symptoms Churg-Strauss syndrome varies greatly from person to person. ... Kidney damage. If Churg-Strauss syndrome affects your kidneys, you can develop glomerulonephritis. ... Diagnosis To diagnose Churg-Strauss syndrome, doctors usually request several types of tests, including: Blood tests. ... Or you may find it helpful to talk to other people with Churg-Strauss syndrome. Preparing for your appointment If you have signs and symptoms common to Churg-Strauss syndrome, make an appointment with your doctor.WG, MPO, PRTN3, IL5, HLA-DRB4, TRBV20OR9-2, RNASE3, HLA-DRB1, TLR2, IL33, TSLP, IL25, MYDGF, CMAS, PTPN22, GCA, PTGDR2, POSTN, CCL26, TNFRSF10C, VCAM1, TNF, THBD, GYPA, FASLG, TRB, FAS, BCHE, PREP, DDR1, MMP9, MMP2, LEPR, LEP, ITGB2, IL10, IL5RA, ACR

-
Ohdo Syndrome
Omim
Lopes and Guion-Almeida (1997) described a case of Ohdo syndrome with the additional features of cleft palate and bladder diverticula. ... Stoll (1999) described a possible case of Ohdo syndrome. White et al. (2003) reported 2 cases of Ohdo syndrome, one with mild and the other with severe features, illustrating the phenotypic variability of the condition. ... Day et al. (2008) examined 8 unrelated patients with the Say-Barber-Biesecker-Young-Simpson type of Ohdo syndrome (SBBYSS; 603736) and reviewed the 25 previously reported patients reported with Ohdo syndrome. One family had a reported recurrence of the syndrome, resulting in pregnancy termination at 20 weeks' gestation. ... Noting that 3 of their patients had thyroid abnormalities, they suggested that Ohdo and SYBBYS syndromes may be the same condition. Clayton-Smith et al. (2011) described SBBYSS as a variant of Ohdo syndrome.
-
Polyostotic Fibrous Dysplasia
Wikipedia
Polyostotic fibrous dysplasia Other names Albright's disease [1] : 578 Specialty Osteology Polyostotic fibrous dysplasia is a form of fibrous dysplasia affecting more than one bone. [2] Fibrous dysplasia is a disorder where bone is replaced by fibrous tissue, leading to weak bones, uneven growth, and deformity. [3] McCune-Albright syndrome includes polyostotic fibrous dysplasia as part of its presentation. [4] When polyostotic fibrous dysplasia manifests in the long bones, limping results; when it manifests in the face, asymmetric growth of the face can result. [3] One treatment that has been used is bisphosphonates . [5] See also [ edit ] Fibrous dysplasia of bone List of radiographic findings associated with cutaneous conditions References [ edit ] ^ James, William; Berger, Timothy; Elston, Dirk (2005). ... Retrieved 2009-02-23 . ^ a b Reference, Genetics Home. "McCune-Albright syndrome" . Genetics Home Reference . Retrieved 2018-10-30 . ^ Lee, Peter A. (5 December 1986). "McCune-Albright Syndrome: Long-term Follow-up". JAMA: The Journal of the American Medical Association . 256 (21): 2980–4. doi : 10.1001/jama.1986.03380210076028 . ... External links [ edit ] Classification D ICD - 10 : Q78.1 ICD - 9-CM : 756.54 MeSH : D005359 DiseasesDB : 7880 External resources Orphanet : 93276 v t e Bone and joint disease Bone Inflammation endocrine : Osteitis fibrosa cystica Brown tumor infection : Osteomyelitis Sequestrum Involucrum Sesamoiditis Brodie abscess Periostitis Vertebral osteomyelitis Metabolic Bone density Osteoporosis Juvenile Osteopenia Osteomalacia Paget's disease of bone Hypophosphatasia Bone resorption Osteolysis Hajdu–Cheney syndrome Ainhum Gorham's disease Other Ischaemia Avascular necrosis Osteonecrosis of the jaw Complex regional pain syndrome Hypertrophic pulmonary osteoarthropathy Nonossifying fibroma Pseudarthrosis Stress fracture Fibrous dysplasia Monostotic Polyostotic Skeletal fluorosis bone cyst Aneurysmal bone cyst Hyperostosis Infantile cortical hyperostosis Osteosclerosis Melorheostosis Pycnodysostosis Joint Chondritis Relapsing polychondritis Other Tietze's syndrome Combined Osteochondritis Osteochondritis dissecans Child leg: hip Legg–Calvé–Perthes syndrome tibia Osgood–Schlatter disease Blount's disease foot Köhler disease Sever's disease spine Scheuermann's_disease arm: wrist Kienböck's disease elbow Panner disease v t e Osteochondrodysplasia Osteodysplasia/ / osteodystrophy Diaphysis Camurati–Engelmann disease Metaphysis Metaphyseal dysplasia Jansen's metaphyseal chondrodysplasia Schmid metaphyseal chondrodysplasia Epiphysis Spondyloepiphyseal dysplasia congenita Multiple epiphyseal dysplasia Otospondylomegaepiphyseal dysplasia Osteosclerosis Raine syndrome Osteopoikilosis Osteopetrosis Other/ungrouped FLNB Boomerang dysplasia Opsismodysplasia Polyostotic fibrous dysplasia McCune–Albright syndrome Chondrodysplasia / chondrodystrophy (including dwarfism ) Osteochondroma osteochondromatosis Hereditary multiple exostoses Chondroma / enchondroma enchondromatosis Ollier disease Maffucci syndrome Growth factor receptor FGFR2 : Antley–Bixler syndrome FGFR3 : Achondroplasia Hypochondroplasia Thanatophoric dysplasia COL2A1 collagen disease Achondrogenesis type 2 Hypochondrogenesis SLC26A2 sulfation defect Achondrogenesis type 1B Autosomal recessive multiple epiphyseal dysplasia Atelosteogenesis, type II Diastrophic dysplasia Chondrodysplasia punctata Rhizomelic chondrodysplasia punctata Conradi–Hünermann syndrome Other dwarfism Fibrochondrogenesis Short rib – polydactyly syndrome Majewski's polydactyly syndrome Léri–Weill dyschondrosteosis This Genodermatoses article is a stub .
-
Coats Plus Syndrome
Medlineplus
Coats plus syndrome is an inherited condition characterized by an eye disorder called Coats disease plus abnormalities of the brain, bones, gastrointestinal system, and other parts of the body. ... Coats plus syndrome and a disorder called leukoencephalopathy with calcifications and cysts (LCC; also called Labrune syndrome) have sometimes been grouped together under the umbrella term cerebroretinal microangiopathy with calcifications and cysts (CRMCC) because they feature very similar brain abnormalities. ... Frequency Coats plus syndrome appears to be a rare disorder. Its prevalence is unknown. Causes Coats plus syndrome results from mutations in the CTC1 gene. ... Researchers are working to determine how telomeres are different in people with CTC1 gene mutations and how these changes could underlie the varied signs and symptoms of Coats plus syndrome. Learn more about the gene associated with Coats plus syndrome CTC1 Inheritance Pattern This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have mutations.

-
Carey Fineman Ziter Syndrome
Wikipedia
Find sources: "Carey Fineman Ziter syndrome" – news · newspapers · books · scholar · JSTOR ( September 2018 ) ( Learn how and when to remove this template message ) Carey Fineman Ziter syndrome Autosomal recessive pattern is the inheritance manner of this condition Specialty Medical genetics Carey Fineman Ziter syndrome is a rare genetic condition. ... Previously diagnosis could be made on clinical features, though brain anomalies could only be determined with an MRI . [4] Differential diagnosis [ edit ] Native American myopathy Moebius syndrome Treatment [ edit ] There is no curative treatment known. ... "Severe congenital myopathy with Möbius, Robin, and Poland sequences: new aspects of the Carey-Fineman-Ziter syndrome". Am J Med Genet A 127A(3):291-293 ^ a b Di Gioia SA, Connors S, Matsunami N, Cannavino J, Rose MF, Gilette NM, Artoni P, de Macena Sobreira NL, Chan WM, Webb BD, Robson CD, Cheng L, Van Ryzin C, Ramirez-Martinez A, Mohassel P, Leppert M, Scholand MB, Grunseich C, Ferreira CR, Hartman T, Hayes IM, Morgan T, Markie DM, Fagiolini M, Swift A, Chines PS, Speck-Martins CE, Collins FS, Jabs EW, Bönnemann CG, Olson EN; Moebius Syndrome Research Consortium, Carey JC, Robertson SP, Manoli I, Engle EC. ... Neurol Genet 4(4):e254. doi: 10.1212/NXG.0000000000000254 ^ "Carey-Fineman-Ziter syndrome" . Orphanet . 2006 . Retrieved 4 August 2018 . ^ Carey JC, Fineman RM, Ziter FA (1982). "The Robin sequence as a consequence of malformation, dysplasia, and neuromuscular syndromes". J Pediatr 101(5):858-864 Classification D OMIM : 254940 MeSH : C536102 DiseasesDB : 33758









