A number sign (#) is used with this entry because of evidence that craniosynostosis-3 (CRS3) is caused by heterozygous mutation in the TCF12 gene (600480) on chromosome 15q21. Description Craniosynostosis is a primary abnormality of skull growth involving premature fusion of the cranial sutures such that the growth velocity of the skull often cannot match that of the developing brain. This produces skull deformity and, in some cases, raises intracranial pressure, which must be treated promptly to avoid permanent neurodevelopmental disability (summary by Fitzpatrick, 2013). Craniosynostosis-3 includes coronal, sagittal, and multisuture forms (Sharma et al., 2013). For discussion of genetic heterogeneity of craniosynostosis, see CRS1 (123100).
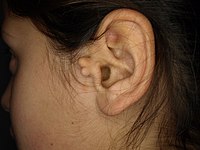
Isolated brachycephaly is a relatively frequent nonsyndromic craniosynostosis consisting of premature fusion of both coronal sutures leading to skull deformity with a broad flat forehead and palpable coronal ridges. Epidemiology Incidence at birth is in the range of 1/20,000. Clinical description The skull deformity is characterized by a short anteroposterior diameter with a compensatory increase in bitemporal width. Supraorbital recession and exorbitism may also be present. Brachycephaly may be associated with facial anomalies (midface hypoplasia, slight hypertelorism and bulging temporal fossae). Increased intracranial pressure (ICP) is frequent and may lead to intellectual deficit if left untreated. In adults, elevated ICP is associated with bony defects in the absence of treatment.
"Changes in severe accidental tetanus mortality in the ICU during two decades in Brazil". Intensive Care Medicine . 28 (7): 930–5. doi : 10.1007/s00134-002-1332-4 .
"The ACR classification criteria for headache disorders in SLE fail to classify certain prevalent headache types". Cephalalgia . 28 (3): 296–9. doi : 10.1111/j.1468-2982.2007.01510.x .
Introduction to Plant Physiology 4th edition ^ "Potassium deficiency in plants" Archived 2010-10-28 at the Wayback Machine , 13 December 2001, 17 November 2010. ^ DEFRA (2010).
"An incidence study on thyroglossal duct cysts in adults". Saudi Medical Journal . 28 (4): 593–7. PMID 17457484 . ^ a b c "Thyroglossal Duct Cysts and Sinuses" .
References [ edit ] ^ a b Welcome to Healthypet.com! Archived 2007-09-28 at the Wayback Machine ^ a b c "Feline Hepatic Lipidosis (Fatty Liver Syndrome)" .
Adult and Pediatric Spine Trauma, An Issue of Neurosurgery Clinics of North America . 28 . Elsevier Health Sciences. ISBN 9780323482844 . ^ Mirghasemi, Alireza; Mohamadi, Amin; Ara, Ali Majles; Gabaran, Narges Rahimi; Sadat, Mir Mostafa (November 2009).
"Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to α-Synuclein Gain" . Cell Metabolism . 28 (4): 605–618.e6. doi : 10.1016/j.cmet.2018.05.019 .
. ^ "The girl who must eat every 15 minutes to stay alive" . The Telegraph . 28 June 2010. Lizzie Velasquez weighs just four stone and has almost zero per cent body fat but she is not anorexic. [...]
A number sign (#) is used with this entry because of evidence that the marfanoid-progeroid-lipodystrophy syndrome (MFLS) is caused by heterozygous mutation occurring in or affecting exon 64 of the FBN1 gene (134797) on chromosome 15q21. Description The marfanoid-progeroid-lipodystrophy syndrome (MFLS) is characterized by congenital lipodystrophy, premature birth with an accelerated linear growth disproportionate to weight gain, and progeroid appearance with distinct facial features, including proptosis, downslanting palpebral fissures, and retrognathia. Other characteristic features include arachnodactyly, digital hyperextensibility, myopia, dural ectasia, and normal psychomotor development (Takenouchi et al., 2013). Takenouchi et al. (2013) noted phenotypic overlap with Marfan syndrome (154700) and Shprintzen-Goldberg craniosynostosis syndrome (182212). Clinical Features Verloes et al. (1998) described a 1-year-old girl with an 'unclassifiable' form of connective tissue disorder.
Progeroid and marfanoid aspect-lipodystrophy syndrome is a rare systemic disease characterized by a neonatal progeroid appearance (not associated with other manifestations of premature aging) associated with facial dysmorphism (e.g. macrocephaly or arrested hydrocephaly, proptosis, downslanting palpebral fissures, retrognathia), generalized, extreme, congenital lack of subcutaneous fat tissue (except in the breast and iliac region) and incomplete signs of Marfan syndrome (mainly severe myopia, joint hyperextensibility and arachnodactyly). Metabolic disturbances are not associated.
A family history of non-reflex epilepsy was obtained in 12% and 28%, respectively, and febrile convulsions (121210) were present in 11% and 7%, respectively.
Hot water reflex epilepsy is a rare neurologic disease characterized by the onset of generalized or focal seizures following immersion of the head in hot water, or with hot water being poured over the head. Primary generalized tonic-clonic seizures have been reported in rare cases.
Von der Werth et al. (2000) directly evaluated 132 family members and detected 28 affected relatives, including 27 who were in the group previously labeled as family history-positive.
Hidradenitis suppurativa, also known as acne inversa, is a chronic skin disease characterized by recurrent boil-like lumps (nodules) under the skin. The nodules become inflamed and painful. They tend to break open (rupture), causing abscesses that drain fluid and pus. As the abscesses heal, they produce significant scarring of the skin. The signs and symptoms of hidradenitis suppurativa appear after puberty, usually in a person's teens or twenties. Nodules are most likely to form in the armpits and groin. They may also develop around the anus, on the buttocks, or under the breasts.