-
Craniofacial-Deafness-Hand Syndrome
Medlineplus
Craniofacial-deafness-hand syndrome is characterized by distinctive facial features, profound hearing loss, and hand abnormalities. The distinctive facial features of people with craniofacial-deafness-hand syndrome result from a variety of developmental abnormalities involving the skull (cranium) and face. ... Frequency Craniofacial-deafness-hand syndrome is an extremely rare condition. ... Causes Craniofacial-deafness-hand syndrome is caused by mutations in the PAX3 gene. ... At least one PAX3 gene mutation has been identified in individuals with craniofacial-deafness-hand syndrome. This mutation appears to affect the ability of the PAX3 protein to bind to DNA.
-
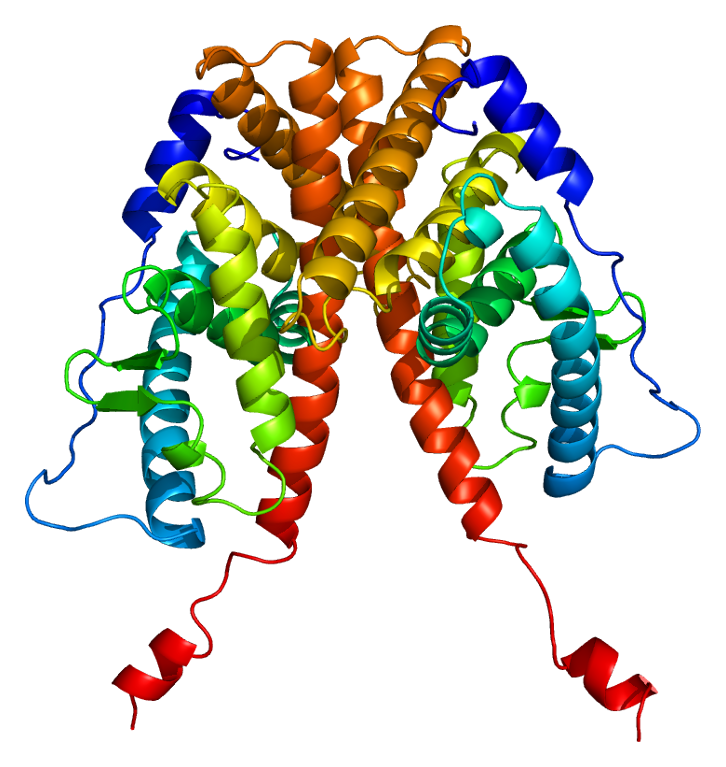
Estrogen Insensitivity Syndrome
Wikipedia
Please help improve it if you can. ( July 2017 ) ( Learn how and when to remove this template message ) Estrogen insensitivity syndrome Other names EIS; Complete estrogen insensitivity syndrome; CEIS [1] EIS results when the function of the estrogen receptor alpha (ERα) is impaired. ... Specialty Endocrinology Estrogen insensitivity syndrome ( EIS ), or estrogen resistance , is a form of congenital estrogen deficiency or hypoestrogenism [2] which is caused by a defective estrogen receptor (ER) – specifically, the estrogen receptor alpha (ERα) – that results in an inability of estrogen to mediate its biological effects in the body. [3] Congenital estrogen deficiency can alternatively be caused by a defect in aromatase , the enzyme responsible for the biosynthesis of estrogens, a condition which is referred to as aromatase deficiency and is similar in symptomatology to EIS. [4] EIS is an extremely rare occurrence. [5] [6] As of 2016, there have been three published reports of EIS, involving a total of five individuals. [6] The reports include a male case published in 1994, [7] [8] a female case published in 2013, [5] [9] and a familial case involving two sisters and a brother which was published in 2016. [6] EIS is analogous to androgen insensitivity syndrome (AIS), a condition in which the androgen receptor (AR) is defective and insensitive to androgens , such as testosterone and dihydrotestosterone (DHT). ... This can be explained by the genetics of each syndrome. AIS is an X-linked recessive condition and thus carried over, by females, into future generations (although the most severe form, complete androgen insensitivity syndrome (CAIS), results in sterility, and hence cannot be passed on to offspring). ... "Congenital estrogen deficiency in men: a new syndrome with different phenotypes; clinical and therapeutic implications in men". ... External links [ edit ] Classification D ICD - 10 : E34.5 OMIM : 300068 External resources Orphanet : 99429 v t e Gonadal disorder Ovarian Polycystic ovary syndrome Premature ovarian failure Estrogen insensitivity syndrome Hyperthecosis Testicular Enzymatic 5α-reductase deficiency 17β-hydroxysteroid dehydrogenase deficiency aromatase excess syndrome Androgen receptor Androgen insensitivity syndrome Familial male-limited precocious puberty Partial androgen insensitivity syndrome Other Sertoli cell-only syndrome General Hypogonadism Delayed puberty Hypergonadism Precocious puberty Hypoandrogenism Hypoestrogenism Hyperandrogenism Hyperestrogenism Postorgasmic illness syndrome Cytochrome P450 oxidoreductase deficiency Cytochrome b5 deficiency Androgen-dependent condition Aromatase deficiency Complete androgen insensitivity syndrome Mild androgen insensitivity syndrome Hypergonadotropic hypogonadism Hypogonadotropic hypogonadism Fertile eunuch syndrome Estrogen-dependent condition Premature thelarche Gonadotropin insensitivity Hypergonadotropic hypergonadism v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions )ESR1, CCDC170, TMEM59, EGFR, BCAR1, BCAR3, AR, PRL, RTN4, SLC39A6, ZFYVE9, TFF3, SHOX, S100B, S100A1, RTN1, PIK3CG, BCL2, PIK3CD, PIK3CB, PIK3CA, NEDD9, INSR, CCN1, GPER1, FOXM1, ERBB3, ERBB2, CDC42, SPECC1
-
Aneuploidy
Wikipedia
The most common aneuploidy that infants can survive with is trisomy 21, which is found in Down syndrome , affecting 1 in 800 births. Trisomy 18 (Edwards syndrome) affects 1 in 6,000 births, and trisomy 13 (Patau syndrome) affects 1 in 10,000 births. 10% of infants with trisomy 18 or 13 reach 1 year of age. [6] Changes in chromosome number may not necessarily be present in all cells in an individual. ... Types [ edit ] key color significance lethal normal male phenotype Klinefelter syndrome (abnormal male) polysomy X and/or Y, (abnormal male) normal female phenotype Turner's syndrome (abnormal female) tetrasomy X, pentasomy X, (abnormal female) Non-autosomal 0 X XX XXX XXXX XXXXX 0 0 X XX XXX XXXX XXXXX Y Y XY XXY XXXY XXXXY XXXXXY YY YY XYY XXYY XXXYY XXXXYY XXXXXYY YYY YYY XYYY XXYYY XXXYYY XXXXYYY XXXXXYYY YYYY YYYY XYYYY XXYYYY XXXYYYY XXXXYYYY XXXXXYYYY YYYYY YYYYY XYYYYY XXYYYYY XXXYYYYY XXXXYYYYY XXXXXYYYYY key color significance case where complete non-mosaic trisomy can never survive to term case where complete non-mosaic trisomy can rarely(barring other complications) survive to term case where complete non-mosaic trisomy can frequently [32] (barring other complications) survive to term Autosomal # monosomy trisomy 1 1p36 deletion syndrome 1q21.1 deletion syndrome Trisomy 1 2 2q37 deletion syndrome Trisomy 2 3 Trisomy 3 4 Wolf–Hirschhorn syndrome Trisomy 4 5 Cri du chat 5q deletion syndrome Trisomy 5 6 Trisomy 6 7 Williams syndrome Trisomy 7 8 Monosomy 8p Monosomy 8q Trisomy 8 9 Alfi's syndrome Kleefstra syndrome Trisomy 9 10 Monosomy 10p Monosomy 10q Trisomy 10 11 Jacobsen syndrome Trisomy 11 12 Trisomy 12 13 Patau syndrome 14 Trisomy 14 15 Angelman syndrome Prader–Willi syndrome Trisomy 15 16 Trisomy 16 17 Miller–Dieker syndrome Smith–Magenis syndrome Trisomy 17 18 Distal 18q- Proximal 18q- Edwards syndrome 19 Trisomy 19 20 Trisomy 20 21 Down syndrome 22 DiGeorge syndrome Phelan–McDermid syndrome 22q11.2 distal deletion syndrome Cat eye syndrome Trisomy 22 Terminology [ edit ] In the strict sense, a chromosome complement having a number of chromosomes other than 46 (in humans) is considered heteroploid while an exact multiple of the haploid chromosome complement is considered euploid . ... Monosomy of the sex chromosomes (45,X) causes Turner syndrome . 2 Disomy Disomy is the presence of two copies of a chromosome. ... The presence of an extra chromosome 21 , which is found in Down syndrome , is called trisomy 21. Trisomy 18 and Trisomy 13 , known as Edwards syndrome and Patau syndrome , respectively, are the two other autosomal trisomies recognized in live-born humans. ... External links [ edit ] Classification D ICD - 10 : Q90 - Q98 ICD - 9-CM : 758 MeSH : D000782 Aneuploidy Testing Aneuploidy FAQ Genetics of Aneuploids v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22

-
Autism-Facial Port-Wine Stain Syndrome
Orphanet
This syndrome is characterised by the presence of a unilateral angioma on the face and autistic developmental problems characterised by language delay and atypical social interactions. Epidemiology So far, the syndrome has been described in four children. Diagnostic methods The initial diagnosis was Sturge-Weber syndrome (see this term), despite the absence of leptomeningeal angiomatosis which is one of the hallmarks of this disease.
-
Acromegaloid Facial Appearance Syndrome
Orphanet
A rare multiple congenital anomalies/dysmorphic syndrome with a probable autosomal dominant inheritance, characterized by a progressively coarse acromegaloid-like facial appearance with thickening of the lips and intraoral mucosa, large and doughy hands and, in some cases, developmental delay. AFA syndrome appears to be part of a phenotypic spectrum that includes hypertrichotic osteochondrodysplasia, Cantu type and hypertrichosis-acromegaloid facial appearance syndrome.
-
Tricho-Retino-Dento-Digital Syndrome
Orphanet
Tricho-retino-dento-digital syndrome is an autosomal dominant ectodermal dysplasia syndrome, characterized by uncombable hair syndrome (see this term), congenital hypotrichosis and dental abnormalities such as oligodontia (see this term) or hyperdontia, and associated with early-onset cataract, retinal pigmentary dystrophy, and brachydactyly with brachymetacarpia.
-
Aids Dysmorphic Syndrome
Wikipedia
AIDS dysmorphic syndrome , also called HIV embryopathy , is a cluster of facial malformations seen in children with perinatal HIV infection . Its status as a syndrome is disputed by the research community. Common symptoms of perinatal HIV infection include candidiasis , lymphocytic interstitial pneumonitis , hepatosplenomegaly , and lymphadenopathy . [1] References [ edit ] ^ "AIDS Dysmorphic Syndrome - NORD (National Organization for Rare Disorders)" .
-
Radioulnar Synostosis-Developmental Delay-Hypotonia Syndrome
Orphanet
Radioulnar synostosis-developmental delay-hypotonia syndrome, also known as Der Kaloustian-McIntosh-Silver syndrome, is an extremely rare syndrome with synostosis described in about 4 patients to date with clinical manifestations including congenital unilateral radioulnar synostosis, generalized hypotonia, developmental delay, and dysmorphic facial features (long face, prominent nose and ears).
-
Deafness-Hypogonadism Syndrome
Orphanet
This syndrome is characterized by the association of congenital mixed hearing loss with perilymphatic gusher (Gusher syndrome or DFN3; see this term), hypogonadism and abnormal behavior. ... Etiology Inheritance appeared to be X-linked recessive and a microdeletion, encompassing the POU3F4 gene (DFN3 locus), was detected in one of the patients leading to the suggestion that deafness - hypogonadism is a contiguous gene deletion syndrome.
-
12q14 Microdeletion Syndrome
Gard
12q14 microdeletion syndrome is a genetic syndrome caused by a missing piece of chromosome 12. The signs and symptoms depend on the size of the missing piece and the genes involved, but generally include growth delay, short stature and feeding difficulties. When this syndrome is inherited, it is passed on in a dominant pattern.
-
Baraitser-Winter Cerebrofrontofacial Syndrome
Gene_reviews
Baraitser-Winter cerebrofrontofacial (BWCFF) syndrome is a multiple congenital anomaly syndrome characterized by typical craniofacial features and intellectual disability (ID) that ranges from mild (usually in those with normal brain structure) to profound (typically in those with a neuronal migration defect). ... Diagnosis/testing. The diagnosis of BWCFF syndrome is established in a proband with a compatible clinical phenotype and a heterozygous gain-of-function variant in either ACTB or ACTG1. ... This syndrome, of unknown etiology, is characterized by significantly widely spaced eyes. Some affected individuals may have undiagnosed BWCFF. Noonan syndrome . In BWCFF syndrome without brain anomalies, the facial appearance in infancy (when the metopic ridge is absent), in association with a chest deformity and nuchal skinfolds or pterygium colli may falsely lead to a diagnosis of Noonan syndrome. Coloboma has been reported in some individuals with Noonan syndrome. Kabuki syndrome . Individuals with Kabuki syndrome have long palpebral fissures reminiscent of BWCFF syndrome.
-
Psychoorganic Syndrome
Wikipedia
Psychoorganic syndrome occurs during atrophy of the brain, most commonly during presenile and senile age (e.g. ... However, these studies were highly criticized and found biased, causing doubt in the existence of the syndrome. Furthermore, various health organizations had difficulty coming to an agreement on the definition of the syndrome. In 1985, the syndrome was defined and provided clear criteria that could be used by patients and medical professionals to help identify the syndrome and isolate ways of prevention. ... "Otoneurological findings in psycho-organic syndrome caused by industrial solvent exposure". ... "Clinical studies of psychoorganic syndromes among workers with exposure to solvents".
-
Parkinson Plus Syndrome
Wikipedia
Parkinson-plus syndromes Other names Disorders of multiple system degeneration Specialty Neurology Parkinson-plus syndromes ( PPS ) is a group of neurodegenerative [1] diseases featuring the classical features of Parkinson's disease ( tremor , rigidity , akinesia / bradykinesia , and postural instability ) with additional features that distinguish them from simple idiopathic Parkinson's disease (PD). Some consider Alzheimer's disease to be in this group. [2] Parkinson-plus syndromes are either inherited genetically or occur sporadically. [3] Atypical parkinsonism, and other Parkinson-plus syndromes are often difficult to differentiate from PD and each other. ... K.; Das S. K. (2003). "Parkinsonism plus syndrome—a review". Neurol India . 51 (2): 183–188. ... "Loss of Dopamine-D2 Receptor Binding Sites in Parkinsonian Plus Syndromes" . Journal of Nuclear Medicine . 39 (6): 954–960. ... M., & Healy, D. G. (2011). Parkinsonism Plus Syndromes. In O. Hardiman & C. P. Doherty (Eds.), Neurodegenerative Disorders (181-196).
-
Spondyloepiphyseal Dysplasia Congenita
Wikipedia
Spranger: Bone Dysplasias, Urban & Fischer 2002, ISBN 3-437-21430-6 Further reading [ edit ] GeneReviews/NIH/NCBI/UW entry on X-Linked Spondyloepiphyseal Dysplasia Tarda OMIM entries on X-Linked Spondyloepiphyseal Dysplasia Tarda External links [ edit ] Classification D ICD - 10 : Q77.7 ICD - 9-CM : 756.9 OMIM : 183900 MeSH : C535788 DiseasesDB : 29410 External resources eMedicine : orthoped/630 v t e Osteochondrodysplasia Osteodysplasia/ / osteodystrophy Diaphysis Camurati–Engelmann disease Metaphysis Metaphyseal dysplasia Jansen's metaphyseal chondrodysplasia Schmid metaphyseal chondrodysplasia Epiphysis Spondyloepiphyseal dysplasia congenita Multiple epiphyseal dysplasia Otospondylomegaepiphyseal dysplasia Osteosclerosis Raine syndrome Osteopoikilosis Osteopetrosis Other/ungrouped FLNB Boomerang dysplasia Opsismodysplasia Polyostotic fibrous dysplasia McCune–Albright syndrome Chondrodysplasia / chondrodystrophy (including dwarfism ) Osteochondroma osteochondromatosis Hereditary multiple exostoses Chondroma / enchondroma enchondromatosis Ollier disease Maffucci syndrome Growth factor receptor FGFR2 : Antley–Bixler syndrome FGFR3 : Achondroplasia Hypochondroplasia Thanatophoric dysplasia COL2A1 collagen disease Achondrogenesis type 2 Hypochondrogenesis SLC26A2 sulfation defect Achondrogenesis type 1B Autosomal recessive multiple epiphyseal dysplasia Atelosteogenesis, type II Diastrophic dysplasia Chondrodysplasia punctata Rhizomelic chondrodysplasia punctata Conradi–Hünermann syndrome Other dwarfism Fibrochondrogenesis Short rib – polydactyly syndrome Majewski's polydactyly syndrome Léri–Weill dyschondrosteosis v t e Diseases of collagen , laminin and other scleroproteins Collagen disease COL1 : Osteogenesis imperfecta Ehlers–Danlos syndrome, types 1, 2, 7 COL2 : Hypochondrogenesis Achondrogenesis type 2 Stickler syndrome Marshall syndrome Spondyloepiphyseal dysplasia congenita Spondyloepimetaphyseal dysplasia, Strudwick type Kniest dysplasia (see also C2/11 ) COL3 : Ehlers–Danlos syndrome, types 3 & 4 Sack–Barabas syndrome COL4 : Alport syndrome COL5 : Ehlers–Danlos syndrome, types 1 & 2 COL6 : Bethlem myopathy Ullrich congenital muscular dystrophy COL7 : Epidermolysis bullosa dystrophica Recessive dystrophic epidermolysis bullosa Bart syndrome Transient bullous dermolysis of the newborn COL8: Fuchs' dystrophy 1 COL9: Multiple epiphyseal dysplasia 2, 3, 6 COL10: Schmid metaphyseal chondrodysplasia COL11: Weissenbacher–Zweymüller syndrome Otospondylomegaepiphyseal dysplasia (see also C2/11 ) COL17: Bullous pemphigoid COL18: Knobloch syndrome Laminin Junctional epidermolysis bullosa Laryngoonychocutaneous syndrome Other Congenital stromal corneal dystrophy Raine syndrome Urbach–Wiethe disease TECTA DFNA8/12, DFNB21 see also fibrous proteinsCOL2A1, CCN6, HAPLN1, HSPG2, CHST3, TRAPPC2, ACAN, CD38, COMP, GLB1, SULT2A1, ZAP70, SMARCAL1, TRPV4, CANT1

-
Aminopterin Syndrome Sine Aminopterin
Omim
Description The pseudoaminopterin syndrome (aminopterin syndrome sine aminopterin; ASSA) is a multiple congenital anomaly disorder characterized by ossification defects of the skull, dysmorphic facial features, delayed development, and variable limb defects. ... Fraser et al. (1987) suggested that this constellation of findings represented a new syndrome, which could be called the aminopterin-like syndrome sine aminopterin (ASSA) syndrome. Fraser et al. (1987) suggested that the disorder reported in 3 sibs as a new syndrome by Crane and Heise (1981) may also be the ASSA syndrome; see 218090. ... Barnicoat et al. (1994) noted that these features suggest both Crane-Heise syndrome and aminopterin syndrome sine aminopterin, and may represent part of a spectrum of abnormalities including these 2 conditions. ... They suggested that pseudoaminopterin syndrome would be an appropriate designation.
-
Stormorken Syndrome
Medlineplus
Stormorken syndrome is a rare condition that affects many body systems. ... Other features include lack of a functioning spleen (asplenia), scaly skin (ichthyosis), headaches, and difficulty with reading and spelling (dyslexia). Frequency Stormorken syndrome is a rare disorder. Approximately a dozen cases have been reported in the medical literature. Causes Stormorken syndrome is caused by a mutation in the STIM1 gene. ... The STIM1 gene mutation involved in Stormorken syndrome leads to production of a STIM1 protein that is constantly turned on (constitutively active), which means it continually stimulates calcium ion entry through CRAC channels regardless of ion levels. ... Learn more about the gene associated with Stormorken syndrome STIM1 Inheritance Pattern This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
-
Hand-Foot-Genital Syndrome
Medlineplus
Hand-foot-genital syndrome is a rare condition that affects the development of the hands and feet, the urinary tract , and the reproductive system. ... Many people with hand-foot-genital syndrome have defects in the ureters, which are tubes that carry urine from each kidney to the bladder, or in the urethra, which carries urine from the bladder to the outside of the body. ... About half of males with this disorder have the urethra opening on the underside of the penis (hypospadias). People with hand-foot-genital syndrome are usually able to have children (fertile). ... Causes Mutations in the HOXA13 gene cause hand-foot-genital syndrome. The HOXA13 gene provides instructions for producing a protein that plays an important role in development before birth. ... Mutations in the HOXA13 gene cause the characteristic features of hand-foot-genital syndrome by disrupting the early development of these structures.

-
Dysostosis
Wikipedia
Dysostosis Specialty Medical genetics A dysostosis is a disorder of the development of bone , in particular affecting ossification . [1] Examples include craniofacial dysostosis , Klippel–Feil syndrome , and Rubinstein–Taybi syndrome . ... External links [ edit ] Classification D MeSH : D004413 DiseasesDB : 31424 SNOMED CT : 109420003 External resources Orphanet : 364559 v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum This article about a disease of musculoskeletal and connective tissue is a stub .
-
Dennie–marfan Syndrome
Wikipedia
Dennie–Marfan syndrome Specialty Neurology Dennie–Marfan syndrome is a syndrome in which there is association of spastic paraplegia of the lower limbs and mental retardation in children with congenital syphilis . [1] Both sexes are affected, and the onset of the disease can be acute or insidious, with slow progression from weakness to quadriplegia . Epilepsy , cataract , and nystagmus may also be found. The syndrome was described by Charles Clayton Dennie in 1929, [2] and Antoine Marfan in 1936. [3] References [ edit ] ^ M.D, Mark E. ... Revue Franc Pédiat 1936; 12: 1–16 External links [ edit ] Dennie-Marfan syndrome at Who Named It? v t e Symptoms and signs relating to infectious diseases Bacterial disease syphilis Hutchinson's teeth Hutchinson's triad Westphal's sign Clutton's joints Dennie–Marfan syndrome Viral disease measles Koplik's spots Parasitic disease African trypanosomiasis Winterbottom's sign General Meningism Fever Liebermeister's rule Faget sign v t e Symptoms and signs relating to the nervous system Neurological examination · Cranial nerve examination Central nervous system Head Battle's sign Kernig's sign Macewen's sign Myerson's sign Stroop test Hirano body Other increased intracranial pressure Cushing's triad Lhermitte's sign Charcot's neurologic triad Peripheral nervous system Reflexes Combination Jendrassik maneuver Legs Plantar reflex Chaddock reflex Oppenheim's sign Westphal's sign Arms Hoffmann's sign Other Arms Froment's sign carpal tunnel syndrome Tinel sign Phalen maneuver Legs Gowers' sign Hoover's sign Lasègue's sign Trendelenburg's sign Torso Beevor's sign General Pain stimulus This article about a medical condition affecting the nervous system is a stub .
-
Van Wyk And Grumbach Syndrome
Wikipedia
Van Wyk and Grumbach syndrome Specialty Endocrine Van Wyk and Grumbach syndrome is a medical condition defined by a combination of hypothyroidism , precocious puberty (usually with delayed bone age) and ovarian cysts in pre- and post-pubertal girls. Contents 1 Presentation 2 Mechanism 3 Diagnosis 4 Treatment 5 History 6 References Presentation [ edit ] Symptoms are ascites , pleural and pericardial effusions, elevated ovarian tumour markers, enlarged pituitary gland and elevated prolactin and alpha-fetoprotein levels. [ citation needed ] Mechanism [ edit ] The presumed pathogenesis is that primary hypothyroidism causes enlargement and hyperstimulation of the pituitary gland which in turn cause ovarian hyperstimulation, ovarian cysts and precocious puberty. [ citation needed ] Diagnosis [ edit ] Diagnosis is made by imaging/sonography and thyroid hormone tests. [ citation needed ] Treatment [ edit ] The syndrome usually responds well to thyroid hormone replacement with complete resolution of symptoms. [ citation needed ] History [ edit ] The syndrome was described in 1960 by Van Wyk and Melvin M. ... P. (2008). "Van Wyk and Grumbach syndrome revisited: Imaging and clinical findings in pre- and postpubertal girls". ... "Elevated alpha-fetoprotein levels in Van Wyk-Grumbach syndrome: A case report and review of literature". ... "Ovarian tumor in a 12-year old female with severe hypothyroidism: A case of Van Wyk and Grumbach syndrome". Pediatric Blood & Cancer . 52 (5): 677–9. doi : 10.1002/pbc.21920 .



