Patients that do not improve stability during physical therapy or develop an increase in pain will be recommended for surgery. [28] References [ edit ] ^ MedlinePlus Encyclopedia : Posterior cruciate ligament (PCL) injury ^ Jonathan Cluett, M.D. (2003-08-05). ... "Posterior Cruciate Ligament Injuries of the Knee Joint". Sports Medicine . 28 (6): 429–41. doi : 10.2165/00007256-199928060-00005 .
. ^ Williams, Kenneth C.; Burdo, Tricia H. (2017-03-28). "HIV and SIV infection - the role of cellular restriction and immune responses in viral replication and pathogenesis" . ... "Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management" . Clinical Microbiology Reviews . 28 (3): 603–661. doi : 10.1128/CMR.00134-14 .
In most cases, if surgery is unsuccessful, severe asphyxia results in the death of the neonate, on average two days after birth. [11] The longest survival ever reported was six years. [28] See also [ edit ] Children's Hospital of Illinois Transplant surgery on Hannah Warren References [ edit ] ^ a b c d "Tracheal agenesis | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . ... "Tracheal agenesis". The Annals of Thoracic Surgery . 28 (3): 295–9. doi : 10.1016/S0003-4975(10)63123-2 .
Tracheal agenesis is a rare birth defect in which the trachea (windpipe) is completely absent (agenesis) or significantly underdeveloped (atresia). Signs and symptoms include polyhydramnios during pregnancy and respiratory distress, bluish skin color ( cyanosis ) and no audible cry shortly after birth. The underlying cause of tracheal agenesis is currently unknown. Approximately 90% of cases are associated with other anomalies, including those of the cardiovascular system, the gastrointestinal system and the genitourinary tract. Some cases may be part of a very rare condition known as VACTERL association . Surgery to repair the trachea may be attempted; however, the long-term outlook is generally poor in most cases.
Tracheal agenesis (TA) is a rare congenital malformation in which the trachea may be completely absent (agenesis), or partially in place but underdeveloped (atresia). In both cases, proximal-distal communication between the larynx and the alveoli of the lungs is lacking. Epidemiology The prevalence at birth is approximately 1 in 50 000. There is a male predominance and an association with prematurity and polyhydramnios. Clinical description Associated congenital malformations are present in 90% of cases, most frequently affecting the cardiovascular or gastro-intestinal systems and the genito-urinary tract, thus overlapping with the much commoner condition, VATER association. Etiology No risk factor for the occurrence of this malformation has ever been suggested.
Cases involving difficult to remove or multiple tumors have responded well to strontium-90 radiotherapy as an alternative to surgery. [28] The prognosis for solitary skin tumors is good, but guarded for tumors in other organs. ... "Recurrence rate, clinical outcome, and cellular proliferation indices as prognostic indicators after incomplete surgical excision of cutaneous grade II mast cell tumors: 28 dogs (1994–2002)". J Vet Intern Med . 20 (4): 933–40. doi : 10.1892/0891-6640(2006)20[933:RRCOAC]2.0.CO;2 .
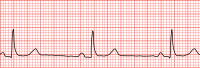
You can help by adding to it . ( December 2018 ) The US Centers for Disease Control and Prevention reported in 2011 that 15.2% of adult males and 6.9% of adult females had clinically-defined bradycardia (a resting pulse rate below 60 BPM). [16] Society and culture [ edit ] Records [ edit ] Daniel Green holds the world record for the slowest heartbeat in a healthy human, with a heart rate measured in 2014 of 26 BPM. [17] Martin Brady holds the Guinness world record for the slowest heart rate with a certified rate over a minute duration of 27 BPM. [18] Professional cyclist Miguel Indurain had, during his career, a resting heart rate of 28 BPM. [19] See also [ edit ] Bezold–Jarisch reflex References [ edit ] ^ "Types of Arrhythmia" . 1 July 2011. ... "The Mammalian Diving Response: An Enigmatic Reflex to Preserve Life?" . Physiology . 28 (5): 284–297. doi : 10.1152/physiol.00020.2013 .
Overview Bradycardia (brad-e-KAHR-dee-uh) is a slow heart rate. The hearts of adults at rest usually beat between 60 and 100 times a minute. If you have bradycardia, your heart beats fewer than 60 times a minute. Bradycardia can be a serious problem if the heart rate is very slow and the heart can't pump enough oxygen-rich blood to the body. If this happens, you may feel dizzy, very tired or weak, and short of breath. Sometimes bradycardia doesn't cause symptoms or complications. A slow heart rate isn't always a concern.
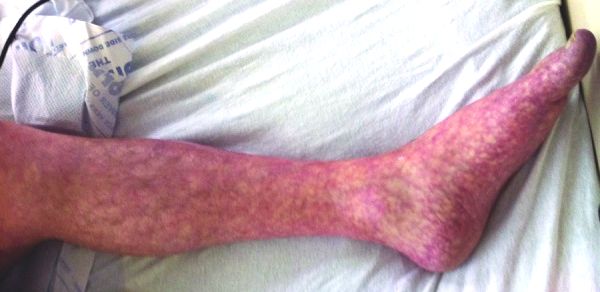
It may be mild, but ulceration may occur later in the summer. [7] Secondary livedo reticularis: Vasculitis autoimmune conditions : Livedoid vasculitis – with painful ulceration occurring in the lower legs Polyarteritis nodosa Systemic lupus erythematosus [8] Dermatomyositis Rheumatoid arthritis Lymphoma Pancreatitis [9] Chronic pancreatitis [10] Tuberculosis Drug-related: Adderall (side effect) [ citation needed ] Amantadine (side effect) Bromocriptine (side effect) Beta interferon treatment, e.g. in multiple sclerosis [11] [12] [13] Livedo reticularis associated with rasagiline [14] Methylphenidate and dextroamphetamine -induced peripheral vasculopathy [15] Gefitinib [16] Obstruction of capillaries: Cryoglobulinaemia – proteins in the blood that clump together in cold conditions [17] Antiphospholipid syndrome due to small blood clots Hypercalcaemia (raised blood calcium levels which may be deposited in the capillaries) Haematological disorders of polycythaemia rubra vera or thrombocytosis (excessive red cells or platelets) Infections (infective endocarditis, syphilis , tuberculosis, Lyme disease) Associated with acute kidney injury due to cholesterol emboli status after cardiac catheterization Arteriosclerosis ( cholesterol emboli ) [18] [19] and homocystinuria (due to Chromosome 21 autosomal recessive Cystathionine beta synthase deficiency ) Intra-arterial injection (especially in drug addicts) Ehlers-Danlos syndrome – connective tissue disorder, often with many secondary conditions, may be present in all types Pheochromocytoma [20] Livedoid vasculopathy and its association with factor V Leiden mutation [21] FILS syndrome (polymerase ε1 mutation in a human syndrome with facial dysmorphism, immunodeficiency, livedo, and short stature) [22] Primary hyperoxaluria, oxalosis (oxalate vasculopathy) [23] [24] [25] [26] [27] Cytomegalovirus infection (very rare clinical form, presenting with persistent fever and livedo reticularis on the extremities and cutaneous necrotizing vasculitis of the toes) [28] Generalized livedo reticularis induced by silicone implants for soft tissue augmentation [29] As a rare skin finding in children with Down syndrome [30] [31] Idiopathic livedo reticularis with polyclonal IgM hypergammopathy [32] CO 2 angiography (rare, reported case) [33] A less common skin lesion of Churg-Strauss syndrome [34] Erythema nodosum-like cutaneous lesions of sarcoidosis showing livedoid changes in a patient with sarcoidosis and Sjögren's syndrome [35] Livedo vasculopathy associated with IgM antiphosphatidylserine-prothrombin complex antibody [36] Livedo vasculopathy associated with plasminogen activator inhibitor-1 promoter homozygosity and prothrombin G20210A heterozygosity [36] As a first sign of metastatic breast carcinoma (very rare) [37] Livedo reticularis associated with renal cell carcinoma (rare) [38] Buerger's disease (as an initial symptom) [39] As a rare manifestation of Graves hyperthyroidism [40] Associated with pernicious anaemia [41] Moyamoya disease (a rare, chronic cerebrovascular occlusive disease of unknown cause, characterized by progressive stenosis of the arteries of the circle of Willis leading to an abnormal capillary network and resultant ischemic strokes or cerebral hemorrhages) [42] Associated with the use of a midline catheter [43] Familial primary cryofibrinogenemia. [44] Diagnosis [ edit ] Livedo reticularis is diagnosed by its clinical appearance and history. ... Clinical and Experimental Dermatology . 28 (4): 452. doi : 10.1046/j.1365-2230.2003.01285_5.x .
Livedo racemosa Specialty Dermatology Livedo racemosa is a skin condition with persistent red or violet discoloration, characterised by a broken, branched, discontinuous and irregular pattern. It can be restricted to the limbs or diffuse. It is usually the first sign of a systemic vascular disorder . [1] See also [ edit ] Livedo reticularis Livedoid dermatitis List of cutaneous conditions References [ edit ] ^ "Livedo racemosa: Clinical, laboratory and histopathological findings in 33 patients and literature review" . Journal of the American Academy of Dermatology . 76 (6): AB425. June 2017. doi : 10.1016/j.jaad.2017.06.143 . ISSN 0190-9622 . This dermatology article is a stub . You can help Wikipedia by expanding it . v t e
December 23, 2014. Archived from the original on 28 June 2015 . Retrieved 23 June 2015 . ^ a b c d e f g h i j k l m n o p q r s t Biggs, WS; Demuth, RH (15 October 2011). ... Challenging the Medicalization of Women's Bodies Archived 2011-06-28 at the Wayback Machine Journal article by Alia Offman, Peggy J.
They found reports of neurologic symptoms in 28 of 110 patients: intermittent episodes of numbness and tingling and sometimes burning sensations in the hands and sometimes the feet, with no obvious precipitant. ... In a further study of 29 additional families, 28 of them ascertained on the basis of NPS, glaucoma was present in 9 (31%).
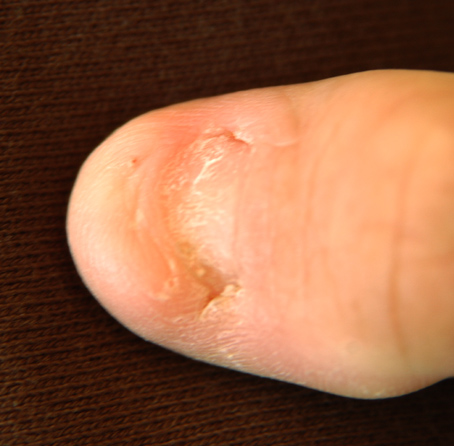
Nail-patella syndrome causes changes in the nails, elbows, kneecaps (patellae), and hip bone. The most common symptom of the syndrome is having missing or underdeveloped fingernails and toenails. Other symptoms may include having small or missing kneecaps, underdeveloped elbows, and an extra small piece of bone on both sides of the hip (called iliac horns). People with nail-patella syndrome are at an increased risk for developing high fluid pressure in the eye (glaucoma) and kidney disease. Nail-patella syndrome is caused by genetic changes (pathogenic variants or mutations) in the LMX1B gene.
Nail–patella syndrome Other names NPS Nail of a patient with nail–patella syndrome Specialty Medical genetics Nail–patella syndrome is a genetic disorder that results in small, poorly developed nails and kneecaps, but can also affect many other areas of the body, such as the elbows, chest, and hips. The name "nail–patella" can be very misleading because the syndrome often affects many other areas of the body, including even the production of certain proteins. [1] : 666 Those affected by NPS may have one or more affected areas of the body, and its severity varies depending on the individual. It is also referred to as iliac horn syndrome , hereditary onychoosteodysplasia ( HOOD syndrome ), Fong disease or Turner–Kieser syndrome . [2] Diagnosis of NPS can be made at birth, but is common for it to remain undiagnosed for several generations. While there is no cure available for NPS, treatment is available and recommended. Contents 1 Signs and symptoms 2 Genetics 3 Diagnosis 4 Treatment 5 See also 6 References 7 External links Signs and symptoms [ edit ] The skeletal structures of individuals who have this disorder may have pronounced deformities.
A rare hereditary patellar dysostosis characterized by nail hypoplasia or aplasia, aplastic or hypoplastic patellae, elbow dysplasia, and the presence of iliac horns as well as renal and ocular anomalies. Epidemiology The reported prevalence is 1/50,000; however, epidemiological studies are lacking. Clinical description Nail-patella syndrome (NPS) is a multisystemic disorder characterized by significant inter- and intrafamilial variability in clinical manifestations and severity of the disease. Cases can range from mild with no functional impact to severe leading to disability. The classical tetrad involves nails dysplasia, absent or hypoplastic patellae, presence of iliac horns, and elbow deformities.
Summary Clinical characteristics. Nail-patella syndrome (NPS) (previously referred to as Fong's disease), encompasses the classic clinical tetrad of changes in the nails, knees, and elbows, and the presence of iliac horns. Nail changes are the most constant feature of NPS. Nails may be absent, hypoplastic, or dystrophic; ridged longitudinally or horizontally; pitted; discolored; separated into two halves by a longitudinal cleft or ridge of skin; and thin or (less often) thickened. The patellae may be small, irregularly shaped, or absent. Elbow abnormalities may include limitation of extension, pronation, and supination; cubitus valgus; and antecubital pterygia. Iliac horns are bilateral, conical, bony processes that project posteriorly and laterally from the central part of the iliac bones of the pelvis. Renal involvement, first manifest as proteinuria with or without hematuria, occurs in 30%-50% of affected individuals; end-stage renal disease occurs up to 15% of affected individuals.
Wenstrup et al. (2002) performed a prospective cohort study on 71 consecutive EDS patients. Twenty of 71, or 28%, had aortic root dilatation defined as greater than 2 standard deviations above population-based norms. ... Mild myopathic features were seen on muscle biopsy of 5 (28%) of 18 patients. Patients with the hypermobility type EDS caused by TNXB haploinsufficiency were least affected.
The large number of distinct types of the Ehlers-Danlos syndrome that have already been identified indicates great heterogeneity, but clearly that heterogeneity is not exhausted by the present classification. Some of the unclassified families are apparently recessive (see 225320); some, such as that reported by Friedman and Harrod (1982), are seemingly dominant. These authors reported a mother and son with large hernias, positional foot deformities, thoracic deformity, asthma, and eczematoid dermatitis. Both had facial asymmetry, prominent nasal bridge and small jaw. The mother had severe thoracolumbar kyphoscoliosis and 'cigarette paper' scars over the legs. She died of dissecting aortic aneurysm and at autopsy had cystic medial necrosis of the aorta and myxomatous degeneration and elongation of the mitral and tricuspid valves.
A rare inherited connective tissue disorder characterized by skin hyperextensibility, widened atrophic scars, and generalized joint hypermobility. Epidemiology Worldwide prevalence is estimated at 1/20,000. Clinical description Skin hyperextensilibity, atrophic scarring, and generalized joint hypermobility are the hallmarks of classical Ehlers-Danlos syndrome (cEDS). However, the clinical picture variably involves multiple organ systems, and clinical presentation may occur anywhere between birth and childhood. In childhood, bruising, skin fragility, and abnormal scarring are common signs. Primary muscular hypotonia may occur and, alongside hypermobility, may delay motor development.
Classical Ehlers-Danlos syndrome (EDS) is a genetic connective tissue disorder that is caused by defects in a protein called collagen . Common symptoms include skin hyperextensibility , abnormal wound healing, and joint hypermobility . More than 90% of people with classical EDS have mutations in COL5A1 or COL5A2 , two genes which encode type V collagen. In rare cases, mutations in the gene encoding type I collagen, COL1A1 gene, may be found. The condition is inherited in an autosomal dominant manner. Treatment and management is focused on preventing serious complications and relieving associated symptoms.
A number sign (#) is used with this entry because Ehlers-Danlos syndrome classic type 2 (EDSCL2) is caused by heterozygous mutation in the collagen alpha-2(V) gene (COL5A2; 120190) on chromosome 2q31. Rarely, specific mutations in the COL1A1 gene (e.g., R134C, 120150.0059) cause classic EDS. Description The Ehlers-Danlos syndromes (EDS) are a group of heritable connective tissue disorders that share the common features of skin hyperextensibility, articular hypermobility, and tissue fragility. The main features of classic Ehlers-Danlos syndrome are loose-jointedness and fragile, bruisable skin that heals with peculiar 'cigarette-paper' scars (Beighton, 1993). There are both severe and mild forms of classic EDS, previously designated EDS I and EDS II, respectively.
This can accompany a variety of disorders. [26] One form of CFD is due to loss-of-mutations in folate receptor-α , (FRα), which transports folates via an endocytic process. [27] [28] [29] While PCFT is expressed primarily at the basolateral membrane of the choroid plexus, FRα is expressed primarily at the apical brush-border membrane. [30] Unlike subjects with HFM, patients with CFD present with neurological signs a few years after birth. ... "Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption". Cell . 127 (5): 917–28. doi : 10.1016/j.cell.2006.09.041 .
A number sign (#) is used with this entry because hereditary folate malabsorption is caused by homozygous or compound heterozygous mutation in the SLC46A1 gene (611672) on chromosome 17q11. Description Hereditary folate malabsorption is an autosomal recessive disorder characterized by signs and symptoms of folate deficiency that appear within a few months after birth. Infants exhibit low blood and cerebrospinal fluid folate levels with megaloblastic anemia, diarrhea, immune deficiency, infections, and neurologic deficits. Treatment with folate supplementation results in resolution of the signs and symptoms. The disorder is caused by impaired intestinal folate absorption and impaired transport of folate into the central nervous system (summary by Qiu et al., 2006).
Hereditary folate malabsorption (HFM) is an inherited disorder of folate transport characterized by a systemic and central nervous system (CNS) folate deficiency manifesting as megaloblastic anemia, failure to thrive, diarrhea and/or oral mucositis, immunologic dysfunction and neurological disorders. Epidemiology The prevalence is unknown. Approximately 30 cases have been reported to date. Clinical description Disease onset usually occurs a few months after birth. Manifestations include failure to thrive, diarrhea and/or mouth ulcers, various neurological manifestations (motor impairment, seizures, developmental delay, cognitive and behavioral disorders), megaloblastic anemia and hypoimmunoglobulinemia. Megaloblastic anemia is the primary manifestation of HFM and can be very severe if untreated.
Hereditary folate malabsorption is a disorder that interferes with the body's ability to absorb certain B vitamins (called folates) from food. Folates are important for many cell functions, including the production of DNA and its chemical cousin, RNA. Infants with hereditary folate malabsorption are born with normal amounts of folates in their body because they obtain these vitamins from their mother's blood before birth. They generally begin to show signs and symptoms of the disorder within the first few months of life because their ability to absorb folates from food is impaired. Infants with hereditary folate malabsorption experience feeding difficulties, diarrhea, and failure to gain weight and grow at the expected rate (failure to thrive).
Summary Clinical characteristics. Hereditary folate malabsorption (HFM) is characterized by folate deficiency with impaired intestinal folate absorption and impaired folate transport into the central nervous system. Findings include poor feeding, failure to thrive, and anemia. There can be leukopenia and thrombocytopenia, diarrhea and/or oral mucositis, hypoimmunoglobulinemia, and other immunologic dysfunction resulting in infections, most often Pneumocystis jerovicii pneumonia. Neurologic manifestations include developmental delays, cognitive and motor impairment, behavioral disorders and, frequently, seizures. Diagnosis/testing. The diagnosis of HFM is established in a proband: with anemia, impaired absorption of an oral folate load, and low cerebrospinal fluid (CSF) folate concentration (even after correction of the serum folate concentration); and/or by the identification of biallelic pathogenic variants in SLC46A1 on molecular genetic testing. Management. Treatment of manifestations: Parenteral (intramuscular) or high-dose oral 5-formyltetrahydrofolate (5-formylTHF, folinic acid, Leucovorin ® ) or the active isomer of 5-formylTHF (Isovorin ® or Fusilev ® ) can obviate the signs and symptoms of HFM.
"Phenomenology and classification of dystonia: a consensus update" . Movement Disorders . 28 (7): 863–873. doi : 10.1002/mds.25475 . ... Madame Curie Bioscience Database . ^ a b c "SGCE gene" . Genetics Home Reference . 2016-03-28 . Retrieved 2016-04-07 . ^ Rudolph, Uwe; Möhler, Hanns (1 February 2006).
Myoclonus-dystonia is a movement disorder that typically affects the neck, torso, and arms. Individuals with this condition experience quick, involuntary muscle jerks or twitches (myoclonus). About half of individuals with myoclonus-dystonia develop dystonia, which is involuntary tensing of various muscles that causes unusual positioning. In myoclonus-dystonia, dystonia often affects one or both hands, causing writer's cramp, or the neck, causing the head to turn (torticollis). The movement problems usually first appear in childhood or early adolescence with the development of myoclonus.
Myoclonus-dystonia syndrome (MDS) is a rare movement disorder characterized by mild to moderate dystonia along with 'lightning-like' myoclonic jerks. Epidemiology The estimated prevalence of MDS in Europe is 1/500,000. Clinical description Disease onset usually occurs in the first or second decade of life. Myoclonus is usually the presenting manifestation and is described as swift ''lightning-like'' jerks that can rarely appear at rest but that are usually triggered by complex motor tasks such as drawing and writing. These movements mainly affect the neck, arms and trunk but can also rarely be seen in the legs or the larynx. In two thirds of cases, dystonia is also experienced in the form of focal or cervical dystonia (see these terms), which may be only mild and does not exacerbate with time.
A number sign (#) is used with this entry because of evidence that myoclonic dystonia-26 (DYT26) is caused by heterozygous mutation in the KCTD17 gene (616386) on chromosome 22q12. Description Myoclonic dystonia-26 is an autosomal dominant neurologic disorder characterized by onset of myoclonic jerks affecting the upper limbs in the first or second decade of life. The disorder is progressive, and patients later develop dystonia with predominant involvement of the craniocervical regions and sometimes the trunk and/or lower limbs. Dystonia dominates the clinical picture (summary by Mencacci et al., 2015). Clinical Features Mencacci et al. (2015) reported 2 unrelated families with myoclonic dystonia.
Nygaard et al. (1999) mapped the locus for myoclonic dystonia in the large American kindred of European and Native American ancestry studied by them to a 28-cM interval spanning the markers D7S802 and D7S1799 in 7q21-q31.
Retrieved 2010-06-16 . [ failed verification ] ^ "Allergic Contact Dermatitis" . Archived from the original on 28 June 2010 . Retrieved 2010-06-16 . ^ a b "Balsam of Peru contact allergy" . Dermnetnz.org. December 28, 2013 . Retrieved March 5, 2014 . ^ Gottfried Schmalz; Dorthe Arenholt Bindslev (2008).
If the tumor is small enough, it might appear at the delivery unpredicted or even later on, in a child. [16] For example, in one case, Epignathus was detected at week 28, which caused change in the structure of face and airways, and the child was born in week 34 with Caesarean section and needed assistance for breathing. [18] If the diagnosis of epignathus does not happen during pregnancy, and the baby survives to birth, even though it becomes immediately apparent, there is a low chance for survival. [8] Other clinical features include dyspnoea , cyanosis , and difficulty in breathing, sucking and swallowing due to the presence of the tumor. [8] Treatment [ edit ] The main priority for treating epignathus is to establish a usable airway free of obstruction and then to feed the baby. [19] [20] This is frequently difficult because there are often complications due to the large mass of the tumor, its location, the complex progression and required corrective modifications. [1] These tumors, characterized as unusual masses or lumps of tissue, are often the result of abnormal tissue growth and may remain localized in one area or spread to other parts of the body. ... "Giant Epignathus Teratoma Discovered at Birth: A Case Report and 7-Year Follow-Up" . Brazilian Dental Journal . 28 (2): 256–261. doi : 10.1590/0103-6440201701368 .
Epignathus is a very rare and life threatening intraoral teratoma, usually arising from the maxilla, mandible, palate or base of skull and invading the cranium, nasopharynx or oral cavity. Epignathus is more commonly seen in females, and presents with various manifestations (depending on the tumor size) including obstructive polyhydramnios in the prenatal period and dyspnea, cyanosis, cough, difficulty in sucking and swallowing, and rarely vomiting (due to swallowing difficulties) postnatally. When large, they can lead to airway obstruction, asphyxia and death in the neonatal period.
It is most prominently present in the Armenians , Sephardic Jews , Ashkenazi Jews , Mizrahi Jews , Cypriots , Kurds , Turks and Arabs. [3] [12] [13] [27] History [ edit ] A New York City allergist, Sheppard Siegal, first described the attacks of peritonitis in 1945; he termed this "benign paroxysmal peritonitis", as the disease course was essentially benign. [28] Dr Hobart Reimann , working in the American University in Beirut , described a more complete picture which he termed "periodic disease". [29] [30] French physicians Henry Mamou and Roger Cattan described the complete disease with renal complications in 1952. [31] [32] See also [ edit ] List of cutaneous conditions Urticarial syndromes References [ edit ] ^ James W, Berger T, Elston D (2005). ... "Semaine Des Hôpitaux de Paris". La Maladie Périodique . N°28 : 1062–1070. ^ Adwan MH (September 2015).
Familial Mediterranean fever (FMF) is an autoinflammatory disorder characterized by recurrent short episodes of fever and serositis resulting in pain in the abdomen, chest, joints and muscles. Epidemiology FMF is primarily found in the south-eastern Mediterranean area. Populations having a high prevalence (1/200-1/1000) of the disease are non-Ashkenazi Jews, Turks, Armenians and Arabs. It is not considered rare in Italy, Greece or Spain. Clinical description Disease onset usually occurs before the age of 30 with an earlier onset corresponding to a more severe phenotype. FMF can be divided into 2 types: FMF type 1 and 2. Type 1 is characterized by attacks (as often as once a week or every few years) of fever and serositis lasting 1-4 days and resolving spontaneously.
A number sign (#) is used with this entry because autosomal dominant familial Mediterranean fever is associated with heterozygous mutation in the MEFV gene (608107). Homozygous or compound heterozygous mutations in the MEFV gene result in classic familial Mediterranean fever (FMF; 249100), which shows autosomal recessive inheritance. Clinical Features Bergman and Warmenius (1968) described a Swedish family in which 4 individuals had recurrent fever and abdominal pain that persisted for 7 to 14 days. Death from renal failure occurred at ages 19, 21, 33, and 58 years. Renal deposition of amyloid was demonstrated in 2 of the 4 patients in whom postmortem examination was performed. The amyloid was described as perireticular and morphologically identical to that seen in FMF.
Summary Clinical characteristics. Familial Mediterranean fever (FMF) is divided into two phenotypes: type 1 and type 2. FMF type 1 is characterized by recurrent short episodes of inflammation and serositis including fever, peritonitis, synovitis, pleuritis, and, rarely, pericarditis and meningitis. The symptoms and severity vary among affected individuals, sometimes even among members of the same family. Amyloidosis, which can lead to renal failure, is the most severe complication, if untreated. FMF type 2 is characterized by amyloidosis as the first clinical manifestation of FMF in an otherwise asymptomatic individual.
Familial Mediterranean fever is an inherited condition characterized by recurrent episodes of painful inflammation in the abdomen, chest, or joints. These episodes are often accompanied by fever and sometimes a rash or headache. Occasionally inflammation may occur in other parts of the body, such as the heart; the membrane surrounding the brain and spinal cord; and in males, the testicles. In about half of affected individuals, attacks are preceded by mild signs and symptoms known as a prodrome. Prodromal symptoms include mildly uncomfortable sensations in the area that will later become inflamed, or more general feelings of discomfort.
Overview Familial Mediterranean fever (FMF) is a genetic autoinflammatory disorder that causes recurrent fevers and painful inflammation of your abdomen, chest and joints. Familial Mediterranean fever (FMF) is an inherited disorder that usually occurs in people of Mediterranean origin — including those of Jewish, Arab, Armenian, Turkish, North African, Greek or Italian ancestry. But it can affect people in any ethnic group. FMF is typically diagnosed during childhood. While there's no cure for this disorder, you may be able to relieve or even prevent signs and symptoms of FMF by following your treatment plan. Symptoms Signs and symptoms of familial Mediterranean fever usually begin during childhood.
Familial Mediterranean fever is an inherited condition characterized by recurrent episodes of painful inflammation in the abdomen, chest, or joints. These episodes are often accompanied by fever and sometimes a rash or headache. Occasionally inflammation may occur in other parts of the body, such as the heart; the membrane surrounding the brain and spinal cord; and in males, the testicles. In about half of affected individuals, attacks are preceded by mild signs and symptoms known as a prodrome. Prodromal symptoms include mildly uncomfortable sensations in the area that will later become inflamed, or more general feelings of discomfort.
Schwabe et al. (1977) reported 197 patients: 131 Armenians, 11 Ashkenazim, 27 non-Ashkenazi Jews, and 28 others. In an analysis of 1,327 cases from the literature, Meyerhoff (1980) found that 50% were Sephardic, 22% Armenian, 11% Arabian, 7% Turkish, and 5% Ashkenazi.
As of 2020, The Cure Starts Now Foundation had over 40 chapter locations in three countries [27] and was a leading funder of over $14 million in DIPG/DMG research in 15 countries and over 90 trials. [28] Nathaniel "Kayne" Finley, an 18 year old from Florida, was diagnosed with DIPG in November 2016. ... She died of pneumonia, related to her weakened health, on January 28, 1962, aged two. [32] Wikimedia Commons has media related to Diffuse intrinsic pontine glioma .
A rare glial tumor characterized by a highly aggressive, diffusely infiltrative pontine lesion generally occurring in children, affecting local nerve fiber tracts and spreading contiguously to involve adjacent structures, but also metastasizing within the central nervous system. Patients mostly present with a short history of symptoms, typically including the classic triad of multiple cranial neuropathies, long tract signs, and ataxia. Signs and symptoms of increased intracranial pressure may present due to obstructive hydrocephalus. Prognosis is poor and not related to histological grade.
Piccini et al. (2007) reported a patient who presented at age 28 years with delusions and lower limb jerks accompanied by intentional myoclonus and cerebellar ataxia. ... The proband developed memory problems at age 28 years. At age 31, he had mild bilateral limb apraxia, dystonia, mild dementia, and hypometabolism of the temporoparietal association cortex on PET scan.
For a general phenotypic description and a discussion of genetic heterogeneity of Alzheimer disease, see 104300. Mapping Liu et al. (2007) conducted a genome screen of 103 patients with late-onset AD who were ascertained as part of the Genetic Research in Isolated Populations (GRIP) program that was conducted in an isolated population from the southwestern area of the Netherlands. Genealogic information resulted in an extremely large and complex pedigree of 4,645 members. The pedigree was split into 35 subpedigrees to reduce the computational burden of linkage analysis. The strongest evidence for linkage, hlod = 5.20 at marker D1S498, was obtained at chromosome 1q21 (AD13).
Early-onset, autosomal dominant Alzheimer disease is a form of Alzheimer disease (AD) that develops before the age of 65. It is diagnosed in families that have more than one member with AD (usually multiple persons in more than one generation) in which the age of onset is consistently before age 60 and often between the ages of 30 and 60 years. In general, AD is a degenerative disease of the brain that causes gradual loss of memory, judgement, and the ability to function socially. There are three subtypes of early-onset familial AD which are each associated with changes (mutations) in unique genes: (1) Alzheimer disease, type 1 is caused by mutations in the APP gene (2) Alzheimer disease, type 3 is caused by mutations in the PSEN1 gene (3) Alzheimer disease, type 4 is caused by mutations in the PSEN2 gene. All subtypes are inherited in an autosomal dominant manner. There is no cure for AD.
For a general phenotypic description and a discussion of genetic heterogeneity of Alzheimer disease (AD), see 104300. Mapping By genomewide linkage analysis of 466 families with late-onset AD, including 730 affected sib pairs, Pericak-Vance et al. (2000) identified a candidate disease locus on chromosome 9p22.1 (nonparametric maximum lod score of 2.97 at marker D9S741). Analysis of a subset of 199 families in which there was at least 1 autopsy-confirmed AD case yielded a higher lod score of 4.31 at the same marker. Scott et al. (2003) reexamined 437 white AD families included in the original report by Pericak-Vance et al. (2000) by considering age of onset as a covariate. Ordered-subsets analysis included continuous covariates in linkage analysis by rank ordering families by a covariate and summing lod scores.
However, the results did not converge with linkage analysis, suggesting that there is more extensive heterogeneity on chromosome 10 than had been appreciated. Liang et al. (2009) examined 28 genes on chromosome 10 for association with LOAD in a Caucasian case-control cohort of 506 cases and 558 controls.
For a phenotypic description and a discussion of genetic heterogeneity of Alzheimer disease (AD), see 104300. Mapping In a late-onset form of familial Alzheimer disease (AD), Pericak-Vance et al. (1997) identified linkage to a locus on chromosome 12. From a series of multiplex families affected with late-onset AD (at least 60 years) ascertained during the previous 14 years and for which DNA had been obtained, Pericak-Vance et al. (1997) selected a subset of 16 families (with 52 AD patients) to use for a genomewide screen. A second subset of 38 families (with 89 AD patients) was used for a follow-up analysis. Linkage analysis was performed using both genetic model-dependent (lod score) and model-independent methods.
A number sign (#) is used with this entry because of evidence that familial Alzheimer disease-1 (AD1) is caused by mutation in the gene encoding the amyloid precursor protein (APP; 104760) on chromosome 21q. A homozygous mutation in the APP gene with a dominant-negative effect on amyloidogenesis was found in a patient with an early-onset progressive dementia and his affected younger sister (104760.0022). A coding single-nucleotide polymorphism (SNP) in the APP gene (104760.0023) has been shown to have a protective effect against Alzheimer disease. See also APP-related cerebral amyloid angiopathy (CAA; 605714), which shows overlapping clinical and neuropathologic features. Description Alzheimer disease is the most common form of progressive dementia in the elderly.
Clinical Features Leuba et al. (2000) described a Swiss family whose members presented with standard clinical and neuropathologic features of Alzheimer disease (104300) and, in particular, severe neurofibrillary tangle degeneration present in the hippocampus and in several cortical areas, together with a large number of beta-amyloid deposits and senile plaques. The brain pathology of 5 deceased members of this family, from 2 generations, represented a coexisting beta-amyloid and prion protein (PrP; 176640) pathology. Frequent beta-amyloid-positive senile plaques were observed, together with senile plaques stained by the monoclonal antibody against PrP(106-126). In all 5 cases, the cerebral cortex showed spongiform changes, mainly in the superficial layers, with some degree of gliosis. Successive sections showed that both beta-amyloid- and PrP-positive senile plaques were deposited in all layers of the frontal and temporal cortex.
For a phenotypic description and a discussion of genetic heterogeneity of Alzheimer disease (AD), see 104300. Mapping In a linkage study of 272 affected sib pairs (ASPs) with AD who were 60 years of age or older at onset, Blacker et al. (1997) found strong evidence of linkage on chromosomes 20 (lod score = 4.09) and 21 (lod score = 5.9). Because of the large marker spacing in the initial genome scan, the candidate region on chromosome 20 spanned 25 cM, corresponding to chromosomal bands 20p12.2-q11.21. Within this candidate region, 1 gene of particular interest was that encoding cystatin-3 (CST3; 604312), because it is known to be an amyloidogenic protein and is codeposited with the amyloid-beta precursor protein (APP; 104760) in amyloid plaques in the brain of AD patients. Using a covariate-based linkage method, Olson et al. (2001) showed that the APP region on chromosome 21q21 is strongly linked to AD-affected sib pairs of the oldest current age (i.e., age either at last exam or at death) who lacked E4 alleles at the apolipoprotein E (APOE; 107741) locus.
A number sign (#) is used with this entry because of the association between late-onset Alzheimer disease-2 (AD2) and the apolipoprotein E (107741) E4 allele. For a general phenotypic description and a discussion of genetic heterogeneity of Alzheimer disease, see 104300. Clinical Features Using positron emission tomography (PET), Reiman et al. (1996) found that 11 cognitively normal subjects aged 50 to 65 years who were homozygous for the APOE4 allele had reduced glucose metabolism in the same regions of the brain as patients with probable Alzheimer disease. The affected areas included temporal, parietal, posterior cingulate, and prefrontal regions. These findings provided preclinical evidence that the presence of the APOE4 allele is a risk factor for Alzheimer disease.
Early-onset autosomal dominant Alzheimer disease (EOAD) is a progressive dementia with reduction of cognitive functions. EOAD presents the same phenotype as sporadic Alzheimer disease (AD) but has an early age of onset, usually before 60 years old. Epidemiology EOAD represents less than 1% of all cases of AD. Clinical description Initial findings of EOAD are mainly disorders of episodic memory or changes in behavior. The patient is often anosognosic and the diagnosis is therefore carried out with the help of a family member. Neurological signs that can be associated with EOAD are spastic paraparesis, intracerebral hemorrhages, seizures, extrapyramidal syndrome and exceptionally cerebellar ataxia.
A number sign (#) is used with this entry because Alzheimer disease-4 (AD4) is caused by heterozygous mutation in the presenilin-2 gene (PSEN2; 600759) on chromosome 1q42. For a phenotypic description and a discussion of genetic heterogeneity of Alzheimer disease, see 104300. Clinical Features Bird et al. (1988) described 5 German kindreds with an autosomal dominant early-onset form of Alzheimer disease. All families were descendants of a group of immigrants, known as the Volga Germans, who came to the United States between 1870 and 1920. Their ancestors had moved from Germany to the southern Volga region of Russia in the 1760s.
Familial Alzheimer disease (familial AD) is a degenerative disease of the brain that causes gradual loss of memory, judgment, and the ability to function socially. About 25% of all Alzheimer disease is familial (more than 2 people in a family have AD). When Alzheimer disease begins before 60 or 65 years of age (early-onset AD) about 60% of the cases are familial (also known as Early-onset familial AD). These cases appear to be inherited in an autosomal dominant manner. There are three subtypes of early-onset familial AD which are each associated with changes (mutations) in unique genes: (1) Alzheimer disease, type 1 is caused by mutations in the APP gene (2) Alzheimer disease, type 3 is caused by mutations in the PSEN1 gene (3) Alzheimer disease, type 4 is caused by mutations in the PSEN2 gene. The condition known as late-onset familial AD includes only the subtype Alzheimer disease, type 2 and is associated with the APOE *4 allele on chromosome 19.
For a general phenotypic description and a discussion of genetic heterogeneity of Alzheimer disease, see 104300. Mapping Liu et al. (2007) conducted a genome screen of 103 patients with late-onset AD who were ascertained as part of the Genetic Research in Isolated Populations (GRIP) program that was conducted in an isolated population from the southwestern area of the Netherlands. Genealogic information resulted in an extremely large and complex pedigree of 4,645 members. The pedigree was split into 35 subpedigrees to reduce the computational burden of linkage analysis. They found significant evidence of linkage of AD to 3q22-q24 (AD15), in a region of 18 cM from D3S3514 to D3S3626 that reached a maximum hlod of 4.44 at marker D3S1579.
For a general phenotypic description and a discussion of genetic heterogeneity of Alzheimer disease, see 104300. Mapping In a genome screen of individuals from an isolated population from the southwestern area of the Netherlands, ascertained as part of the Genetic Research in Isolated Populations (GRIP) program, Liu et al. (2007) found the strongest evidence of linkage for chromosome 1q21 (AD13; 611152). Approximately 30 cM upstream of this locus, at 1q25, another peak (AD14) was found (hlod = 4.0 at marker D1S218). Liu et al. (2007) noted that these 2 loci were in a linkage region spanning 1q21-q31 identified by Zubenko et al. (1998), Hiltunen et al. (2001), Myers et al. (2002), and Blacker et al. (2003). Haplotype analysis showed that the 2 linkage peaks on chromosome 1q21 and 1q25 are explained by different haplotypes, of 15 cM and 21 cM, respectively, segregating in different families.
For a phenotypic description and a discussion of genetic heterogeneity of Alzheimer disease (AD), see 104300. Mapping Giedraitis et al. (2006) conducted a genome scan with 369 microsatellite markers in 12 extended families collected in Sweden. Age at disease onset ranged from 53 to 78 years, but in 10 of the families there was at least 1 member with age at onset of less than 65 years. Mutations in known early-onset Alzheimer disease susceptibility genes were excluded. All persons were genotyped for APOE (107741), but no clear linkage with the E4 allele was observed.
For a phenotypic description and a discussion of genetic heterogeneity of Alzheimer disease (AD), see 104300. Mapping In an extended multiplex family, ascertained in a population-based study of early-onset AD in the northern Netherlands, Rademakers et al. (2005) obtained conclusive evidence of linkage of AD with a candidate region of 19.7 cM at 7q36. They identified a shared haplotype at 7q36 between the index family and 3 of 6 multiplex AD-affected families from the same geographic region, which was indicative of a founder effect and defined a priority region of 9.3 cM. Mutation analysis of coding exons of 29 candidate genes identified only an exonic silent mutation in the PAXIP1 gene (608254), 38030G-C in the exon 10 genomic sequence, which affected codon 626. It remained to be determined whether PAXIP1 has a functional role in the expression of AD in the index family or whether another mutation at this locus explained the observed linkage and sharing.
For a phenotypic description and a discussion of genetic heterogeneity of Alzheimer disease (AD), see 104300. Mapping In a systematic survey of the human genome in patients with AD, Zubenko et al. (1998) identified D10S1423, located at 10p13, as a candidate susceptibility locus. The allelic associations in this survey were observed in independent samples of autopsied AD cases and controls from geographically disparate sites (Boston and Pittsburgh). Majores et al. (2000) replicated these findings by identifying an association of the D10S1423 234-bp allele with AD in an ethnically homogeneous group of 397 German AD cases and controls. Zubenko et al. (2001) described a prospective, longitudinal, double-blind assessment of the age-specific risk of AD encountered by 325 asymptomatic first-degree relatives of AD probands who carried the D10S1423 234-bp allele, the APOE E4 allele (107741), or both, after 11.5 years of systematic follow-up.
Ellis' family, along with her community, has worked to raise awareness of the disease and helped pass "Cove's Law", which provides parents the option to have prenatal screening for the disease, which can potentially save the child. [26] Other animals [ edit ] Krabbe disease may also be found in cats [27] and in dogs, particularly the West Highland White Terrier and Cairn Terriers . [28] [29] See also [ edit ] Maria Luisa Escolar The Myelin Project The Stennis Foundation References [ edit ] ^ a b c d e Langan, Thomas J (23 November 2016). ... "Hematopoietic Stem Cell Transplantation in Late-Onset Krabbe Disease: No Evidence of Worsening Demyelination and Axonal Loss 4 Years Post-allograft". Journal of Neuroimaging . 28 (3): 252–255. doi : 10.1111/jon.12502 .
Summary Clinical characteristics. Krabbe disease comprises a spectrum ranging from infantile-onset disease (i.e., onset of extreme irritability, spasticity, and developmental delay before age 12 months) to later-onset disease (i.e., onset of manifestations after age 12 months and as late as the seventh decade). Although historically 85%-90% of symptomatic individuals with Krabbe disease diagnosed by enzyme activity alone have infantile-onset Krabbe disease and 10%-15% have later-onset Krabbe disease, the experience with newborn screening (NBS) suggests that the proportion of individuals with possible later-onset Krabbe disease is higher than previously thought. Infantile-onset Krabbe disease is characterized by normal development in the first few months followed by rapid severe neurologic deterioration; the average age of death is 24 months (range 8 months to 9 years). Later-onset Krabbe disease is much more variable in its presentation and disease course. Diagnosis/testing. The two diagnostic scenarios are the following: Scenario 1.
Krabbe disease (also called globoid cell leukodystrophy) is a severe neurological condition. It is part of a group of disorders known as leukodystrophies, which result from the loss of myelin (demyelination) in the nervous system. Myelin is the protective covering around nerve cells that ensures the rapid transmission of nerve signals. Krabbe disease is also characterized by abnormal cells in the brain called globoid cells, which are large cells that usually have more than one nucleus . The most common form of Krabbe disease, called the infantile form, usually begins before the age of 1.
A number sign (#) is used with this entry because Krabbe disease is caused by homozygous or compound heterozygous mutation in the galactosylceramidase gene (GALC; 606890) on chromosome 14q31. Description Krabbe disease is an autosomal recessive lysosomal disorder affecting the white matter of the central and peripheral nervous systems. Most patients present within the first 6 months of life with 'infantile' or 'classic' disease manifest as extreme irritability, spasticity, and developmental delay (Wenger et al., 2000). There is severe motor and mental deterioration, leading to decerebration and death by age 2 years. Approximately 10 to 15% of patients have a later onset, commonly differentiated as late-infantile (6 months to 3 years), juvenile (3 to 8 years), and even adult-onset forms.
Krabbe disease affects the development and function of the nervous system. There are several types of Krabbe disease that differ based on the age that symptoms begin. The early-onset type of Krabbe disease is the most common and the most severe. Babies who have early-onset (infantile) Krabbe disease typically develop features in the first six months of life. Symptoms of infantile Krabbe disease may include irritability, failure to thrive , slowed development, and unexplained fevers.
A number sign (#) is used with this entry because atypical Krabbe disease due to saposin A deficiency is caused by mutation in the prosaposin gene (PSAP; 176801). Krabbe disease (245200) is a genetically distinct disorder caused by mutation in the galactosylceramidase (galactocerebrosidase) gene (GALC; 606890). Clinical Features Spiegel et al. (2005) reported a child, born of consanguineous Arab parents, with saposin A deficiency. She developed normally until age 3.5 months, at which time she showed rapid neurologic deterioration and loss of acquired milestones. At age 6 months, she was almost in a vegetative state without eye contact and with little spontaneous movement.
Krabbe disease is a lysosomal disorder that affects the white matter of the central and peripheral nervous systems. It includes infantile, late-infantile/juvenile and adult forms. Epidemiology It has an estimated prevalence of 1/100,000 in the Northern European population (higher in certain populations) and a worldwide incidence of 1/100,000-1/250,000 live births. The infantile form is the most common form and accounts for 85-90% of cases in the Northern European population. Clinical description The infantile form has an onset at 2-6 months of age and is divided into 3 stages. In the first stage, symptoms include irritability, stiffness, poor head control, feeding difficulties, intermittent thumb clasp, episodes of increased temperature, and developmental delay.