Hyperprolactinemic SAHA syndrome Specialty Dermatology Hyperprolactinemic SAHA syndrome is a cutaneous condition characterized by lateral hairiness, oligomenorrhea, and sometimes acne, seborrhea, FAGA I, and even galactorrhea. [1] See also [ edit ] SAHA syndrome List of cutaneous conditions References [ edit ] ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007).
Sweet's syndrome-like dermatosis Specialty Dermatology Sweet's syndrome-like dermatosis is a cutaneous condition associated with bowel disorders. [1] See also [ edit ] Sweet's syndrome List of cutaneous conditions References [ edit ] ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007).
3q26q27 microdeletion syndrome is a rare partial autosomal monosomy syndrome characterized by neonatal hypotonia, prenatal and postnatal growth deficiency, severe feeding difficulties, global developmental delay and intellectual disability, dental anomalies (delayed tooth eruption, delayed loss of primary teeth, dental crowding), recurrent respiratory infections, thrombocytopenia and facial dysmorphism (flat facial profile, medially sparse eyebrows, epicanthal folds, flat nasal bridge and tip, short philtrum). Behavioral abnormalities (ADHD, Asperger syndrome) have also been reported.
When this pain persists and worsens, doctors call it myofascial pain syndrome. Risk factors Myofascial pain syndrome is caused by a stimulus, such as muscle tightness, that sets off trigger points in your muscles. ... Complications Complications associated with myofascial pain syndrome may include: Sleep problems. Signs and symptoms of myofascial pain syndrome may make it difficult to sleep at night. ... Some research suggests that myofascial pain syndrome may develop into fibromyalgia in some people. ... Some doctors believe myofascial pain syndrome may play a role in starting this process. ... Acupuncture also appears to be helpful for some people who have myofascial pain syndrome. Lifestyle and home remedies Take care of yourself if you have myofascial pain syndrome.
Myofascial pain syndrome Other names Chronic myofascial pain, myofascial pain and dysfunction syndrome (MPDS or MFPDS) Specialty Rheumatology Differential diagnosis Giant cell arteritis , arthritis [1] Myofascial pain syndrome ( MPS ), also known as chronic myofascial pain ( CMP ), is a syndrome characterized by chronic pain in multiple myofascial trigger points ("knots") and fascial (connective tissue) constrictions. ... At least one study rules out trigger points: "The theory of myofascial pain syndrome (MPS) caused by trigger points (TrPs) ... has been refuted. ... Regular, non-intense activity is also encouraged. [12] References [ edit ] ^ a b c d e f "Myofascial Pain Syndrome - Dental Disorders" . Merck Manuals Professional Edition . Retrieved 27 May 2019 . ^ Bennett, Robert (2007). "Myofascial pain syndromes and their evaluation". Best Practice & Research Clinical Rheumatology . 21 (3): 427–45. doi : 10.1016/j.berh.2007.02.014 . PMID 17602992 . ^ Mayo Clinic Staff (3 Dec 2009). "Myofascial pain syndrome: Symptoms" . Retrieved 8 May 2011 . ^ Quintner JL, Bove GM, Cohen ML (2015).
A number sign (#) is used with this entry because Waardenburg syndrome type 3 (WS3) is caused by heterozygous or homozygous mutation in the PAX3 gene (606597) on chromosome 2q36. Waardenburg syndrome type 1 (WS1; 193500) is also caused by heterozygous mutation in the PAX3 gene. ... WS type 3 is also referred to as 'Klein-Waardenburg syndrome' (Gorlin et al., 1976). Clinical Variability of Waardenburg Syndrome Types 1-4 Waardenburg syndrome has been classified into 4 main phenotypes. ... Wollnik et al. (2003) reported a family in which both parents were heterozygous for a Y90H mutation in PAX3 (606597.0013) and had Waardenburg syndrome type 1; the offspring was homozygous for the mutation and had Waardenburg syndrome type 3. ... From these studies, Pasteris et al. (1992) concluded that Waardenburg syndrome type 3 is a contiguous gene syndrome.
Le Merrer et al. (1988) described 2 consanguineous sibships with 3 children and 2 children with this syndrome, respectively. The distal abnormalities of the limbs included syndactyly IV-V, fusion of metacarpals IV-V, absence of the fifth toes, and, in the second family, hypoplasia of the fibula with short femur or tibia. ... The authors stated that these features were consistent with ophthalmoacromelic syndrome (OAS), but noted that this was the first report of 2/3 syndactyly of the fingers, metacarpal polydactyly, or lobster-claw hand deformity in OAS. Tekin et al. (2000) reported an additional patient with ophthalmoacromelic syndrome. The parents were of Turkish ancestry and were reportedly distant relatives. ... Teiber et al. (2007) described a 15-month-old girl with ophthalmoacromelic syndrome. She had an asymmetric face, left unilateral microphthalmia, short, hypoplastic fifth fingers with a single interphalangeal crease, and proximal implantation of the second and third toes. ... Mapping Hamanoue et al. (2009) performed homozygosity mapping in 2 unrelated consanguineous Lebanese families with ophthalmoacromelic syndrome, 1 of which had been previously reported by Megarbane et al. (1998), and identified a 422-kb common region on chromosome 10p11.23 between STS9 and STS12.
Ophthalmo-acromelic syndrome is a condition that results in malformations of the eyes, hands, and feet. ... The most common hand and foot malformation seen in ophthalmo-acromelic syndrome is missing fingers or toes (oligodactyly). ... Frequency The prevalence of ophthalmo-acromelic syndrome is not known; approximately 35 cases have been reported in the medical literature. Causes Mutations in the SMOC1 gene cause ophthalmo-acromelic syndrome. The SMOC1 gene provides instructions for making a protein called secreted modular calcium-binding protein 1 (SMOC-1). ... Some people with ophthalmo-acromelic syndrome do not have an identified mutation in the SMOC1 gene.
Differential diagnosis Differential diagnoses include isolated cryptophthalmia and other forms of syndromic microphthalmia such as microphthalmia, Lenz type, oculofaciocardiodental syndrome and anophthalmia/microphthalmia-esophageal atresia (see these terms).
Group of symptoms caused by obstruction of the superior vena cava Superior vena cava syndrome (Mediastinal syndrome) Other names SVC obstruction [1] Superior vena cava syndrome in a person with bronchogenic carcinoma . ... Retrieved 4 June 2019 . ^ Rice, TW (January 2006). "The superior vena cava syndrome: clinical characteristics and evolving etiology". ... "Superior Vena Cava Syndrome". In Chang, AE; Ganz, PA; Hayes, DF; et al. ... "Clinical practice. Superior vena cava syndrome with malignant causes". N Engl J Med . 356 (18): 1862–9. doi : 10.1056/NEJMcp067190 . ... "Transvenous pacing lead-induced Superior Vena Cava Syndrome: What do we know?". Surgical Practice . 13 (4): 125–126. doi : 10.1111/j.1744-1633.2009.00462.x .
Possibly factitious syndrome Resignation syndrome (also called traumatic withdrawal syndrome or traumatic refusal , in Swedish : Uppgivenhetssyndrom ) is a, possibly factitious, dissociative syndrome that induces a catatonic state , first described in Sweden in the 1990s. ... (November 2009). "Pervasive refusal syndrome as part of the refusal-withdrawal-regression spectrum: critical review of the literature illustrated by a case report" . ... "Comment on the paper "Pervasive Refusal Syndrome (PRS) 21 years on-a reconceptualization and renaming" by Ken Nunn, Bryan Lask and Isabel Owen" . ... PMID 17080781 . ^ "12yo refugee on hunger strike on Nauru suffering from resignation syndrome, doctors say - ABC News (Australian Broadcasting Corporation)" . ... Retrieved 2018-09-25 . ^ O'Connor, Beth. "What is Resignation Syndrome?" . Medicin Sans Frontières - Doctors Without Borders .
Diagnosis VLDLR cerebellar hypoplasia ( VLDLR -CH) is a subgroup of dysequilibrium syndrome (DES), a spectrum of genetically heterogeneous conditions that combines non-progressive cerebellar ataxia with intellectual disability inherited in an autosomal recessive manner. ... Nomenclature VLDLR -CH is a clinically and molecularly well-defined subgroup of dysequilibrium syndrome (DES). Prevalence The actual frequency of VLDLR -CH is unknown. ... Genes and Disorders of Interest in the Differential Diagnosis of VLDLR Cerebellar Hypoplasia View in own window Gene(s) Disorder Brain Imaging Neurologic Findings AHI1 CPLANE1 CC2D2A CEP290 (~34 genes) 1 Joubert syndrome & related disorders 2 "Molar tooth sign" (hypoplasia of cerebellar vermis & assoc brain stem abnormalities resembling a tooth) DD & severe cognitive impairment (in some individuals) Episodic hyperpnea or apnea &/or atypical eye movements Truncal ataxia ALG1 ALG6 PMM2 (~42 genes) 3 Congenital disorders of glycosylation Cerebellar atrophy DD/ID Hypotonia & ataxia Strabismus ATCAY Cayman-type cerebellar ataxia (OMIM 601238) 4 CH Cerebellar ataxia w/wide-based gait Dysarthria Intention tremor DD/ID ATM Ataxia-telangiectasia 5 Cerebellar atrophy (may not be obvious in very young individuals) Choreoathetosis Oculomotor apraxia Progressive cerebellar ataxia beginning at ages 1-4 yrs ATP8A2 CAMRQ4 (OMIM 615268) Cerebellar atrophy Congenital cerebellar ataxia ID CA8 CAMRQ3 (OMIM 613227) Congenital cerebellar ataxia ID EXOSC3 RARS2 SEPSECS TSEN2 TSEN34 TSEN54 VRK1 6 PCH types 1 & 2 (see EXOSC3 -PCH & TSEN54 -PCH) Cerebellar vermis hypoplasia & hypoplasia of the pons (more severe than small pons seen in VLDLR -CH) Progressive motor degeneration (similar to spinal muscular atrophy) in PCH1 Dyskinesia in PCH2 RELN RELN lissencephaly w/CH 7 (OMIM 257320) Cerebellar signs of RELN -LCH that differ from VLDLR -CH: More significant lissencephaly w/anterior>posterior gradient A malformed hippocampus Profound CH w/complete absence of detectable folia Congenital cerebellar ataxia Hypotonia ID Epilepsy Strabismus SACS ARSACS ( a utosomal r ecessive s pastic a taxia of C harlevoix- S aguenay) Atrophy of superior vermis Distal muscle wasting Distal sensorimotor neuropathy (predominant in legs) Dysarthria Early-onset ataxia Extensor plantar reflexes Horizontal gaze-evoked nystagmus Spasticity SIL1 Marinesco-Sjögren syndrome 8 Cerebellar atrophy Cerebellar ataxia Mild-to-severe cognitive impairment Hypotonia & muscle weakness TWNK Infantile-onset spinocerebellar ataxia 9 Atrophy of cerebellum, brain stem, & spinal cord Normal development until age 1 yr, followed by onset of ataxia, muscle hypotonia, loss of deep-tendon reflexes, athetosis, ophthalmoplegia, & sensorineural deafness in childhood 10 Epilepsy can → serious & often fatal encephalopathy. WDR81 CAMRQ2 (OMIM 610185) CH Congenital cerebellar ataxia ID CAMRQ = cerebellar ataxia, mental retardation, and dysequilibrium syndrome; CDG = congenital disorder of glycosylation; CH = cerebellar hypoplasia; DD = developmental delay; ID = intellectual disability; LCH = lissencephaly with cerebellar hypoplasia; PCH = pontocerebellar hypoplasia 1. To date, pathogenic variants in 34 genes are known to cause Joubert syndrome. AHI1 , CPLANE1 , CC2D2A , and CEP290 are some of the most commonly involved genes. 2.
Kowarski syndrome Other names Short stature due to growth hormone qualitative anomaly This condition is inherited in an autosomal recessive manner. ... Kowarski syndrome was assumed to be a very rare disorder (officially recognized as an “orphan disease”). ... In 1995, it was also suggested [9] that some cases of the neurosecretory growth failure syndrome might have the Kowarski syndrome. ... Their study demonstrated a decrease ability of the growth hormone from children with the Kowarski syndrome to bind with living IM-9 cells. ... The study found that the RRA-GH/RIA-GH ratio in NS subjects was normal but significantly below normal (P<0.005) in the Kowarski syndrome patients. The authors proposed the use of their test for the diagnosis of the Kowarski syndrome.
Prevalence is unknown but only a few cases have been reported in the literature. The syndrome is caused by various mutations in the GH1 gene (17q22-q24) that result in structural GH anomalies and a biologically inactive molecule.
Disconnection syndrome can also lead to aphasia , left-sided apraxia , and tactile aphasia, among other symptoms. ... Studies of the monkey brain led to his theory that disconnection syndromes were higher function deficits. ... He described the callosal syndrome, an example of a disconnection syndrome, which is a lesion in the corpus callosum that leads to tactile anomia in just the patient’s left hand. [4] Though Geschwind made significant advances in describing disconnection syndromes, he was not completely accurate. ... "The rises and falls of disconnection syndromes" (PDF) . Brain . 128 (Pt 10): 2224–2239. doi : 10.1093/brain/awh622 . ... "Visualizing the neural bases of a disconnection syndrome with diffusion tensor imaging".
Ortner's syndrome Image of aortic anatomy showing proximity of vagus nerve and its recurrent branch to the aorta Specialty Neurology Ortner's syndrome is a rare cardiovocal syndrome and refers to recurrent laryngeal nerve palsy from cardiovascular disease . [1] It was first described by Norbert Ortner (1865–1935), an Austrian physician, in 1897. ... Due to compression of the recurrent laryngeal nerve, it can cause the hoarseness of the voice, which can also be a sign of mitral stenosis. A second Ortner's syndrome, Ortner's syndrome II, refers to abdominal angina . ... "Painless aortic dissection presenting as hoarseness of voice: cardiovocal syndrome: Ortner's syndrome". The American Journal of Emergency Medicine . 17 (4): 361–3. doi : 10.1016/s0735-6757(99)90087-6 . PMID 10452434 . ^ a b Al Kindi AH, Al Kindi FA, Al Abri QS, Al Kemyani NA (October 2016). "Ortner's syndrome: Cardiovocal syndrome caused by aortic arch pseudoaneurysm" . ... PMID 126321 . ^ a b Hirata M, Sunahori-Watanabe K, Isihara M, Shibuto N, Hiramatsu S, Miyawaki Y, et al. (2 January 2018). "Cardiovocal syndrome (Ortner syndrome) associated with secondary pulmonary arterial hypertension in a patient with mixed connective tissue disease".
A number sign (#) is used with this entry because diffuse leiomyomatosis with Alport syndrome (DL-ATS) represents a contiguous gene deletion syndrome involving deletion of the N-terminal regions of 2 contiguous genes localized in a head-to-head manner on chromosome Xq22: COL4A5 (303630), which is the usual site of mutations in X-linked Alport syndrome (ATS; 301050), and COL4A6 (303631), which regulates smooth muscle differentiation and morphogenesis. Clinical Features Cochat et al. (1988) described the association of diffuse leiomyomatosis with Alport syndrome in 12 patients from 5 pedigrees. ... (See 150700 for a syndrome of leiomyoma of vulva and esophagus; this combination should prompt search for other features of Alport syndrome in the female patient herself or in male relatives.) ... Because of the manifestations, the diagnosis of Alport syndrome with diffuse leiomyomatosis was considered. ... This finding indicated that the association of Alport syndrome with leiomyomatosis is X-linked and probably represents a contiguous gene deletion syndrome involving genes for congenital cataract, diffuse esophageal leiomyomatosis, and Alport syndrome.
It is possible that the disorder in these 2 reports was in fact the leiomyomatosis nephropathy syndrome (308940), an entity in which expression is milder in females, and which appears to be a contiguous gene deletion syndrome (Antignac et al., 1992) that in many cases also includes congenital cataract.
Description Asperger syndrome is considered to be a form of childhood autism (see, e.g., 209850). The DSM-IV (American Psychiatric Association, 1994) specifies several diagnostic criteria for Asperger syndrome, which has many of the same features as autism. ... Gillberg et al. (2001) described the development of the Asperger syndrome (and high-functioning autism) Diagnostic Interview (ASDI), which they claimed has a strong validity in the diagnosis of the disorder. For a discussion of genetic heterogeneity of Asperger syndrome, see ASPG1 (608638). Clinical Features Anneren et al. (1995) reported a 10-year-old boy with Asperger syndrome. ... Mapping By Southern blot analysis of the chromosome 17p13 breakpoints in 2 patients with Asperger syndrome, Tentler et al. (2002) determined that the breakpoint is positioned within a 0.7-kb region located between the CHRNE (100725) and GP1BA (606672) genes, 0.8 kb from the 5-prime end of the CHRNE gene.
A rare lipodystrophic syndrome characterized by loss of adipose tissue, and is a syndrome of insulin resistance that leads to increased cardiovascular risk. ... Clinical description The clinical phenotype is similar to that of Berardinelli-Seip syndrome (see this term), but lipoatrophy appears secondarily during childhood, adolescence or adulthood, and as a result the syndrome is thought to be acquired. ... Three types of the disease have been described: 1) a form with panniculitis (inflammatory nodules followed by lipoatrophy), 2) an autoimmune form that is readily associated with other syndromes such as chronic active hepatitis, Hashimoto struma and hemolytic anemia, but also with dermatomyositis and Sjogren's syndrome (see these terms), 3) idiopathic. ... Differential diagnosis Differential diagnoses include other forms of extreme insulin resistance (Rabson-Mendenhall syndrome, leprechaunism, Berardinelli type lipodystrophy and insulin resistance syndromes types A and B; see these terms) and other lipodystrophies. ... Prognosis The prognosis is not well known but is probably related to cardiovascular risk (linked to the insulin-resistance syndrome) and to the underlying cause of the disease.
Acquired generalized lipodystrophy Other names Lawrence-Seip syndrome Acquired generalized lipodystrophy (also known as "Lawrence syndrome," [1] and "Lawrence–Seip syndrome", [1] abbreviation: AGL) is a rare skin condition that appears during childhood or adolescence, characterized by fat loss affecting large areas of the body, particularly the face, arms, and legs. [2] : 496 There are 4 types of lipodystrophy based on its onset and areas affected: acquired or inherited ( congenital or familial), and generalized or partial . ... Continuous elevation in triglyceride levels further contributes to metabolic problems including insulin resistance . [12] As the level of leptin in the body is proportional to the amount of adipose tissue present, AGL patients also have a deficiency of leptin which contributes to excessive eating and worsens the metabolic syndrome . [12] In a few patients with AGL, the presence of antibodies against adipocyte has been identified. [13] Diagnosis [ edit ] Diagnosis is made comprehensively, together with visual observation, body fat assessment, a review of lab panels consisting of A1c, glucose, lipid, and patient history. ... Retrieved 2017-11-07 . ^ a b c d e f g h i j k Hussain, Iram; Garg, Abhimanyu (2016). "Lipodystrophy Syndromes" . Endocrinology and Metabolism Clinics of North America . 45 (4): 783–797. doi : 10.1016/j.ecl.2016.06.012 . ... "Dysregulation of insulin-like growth factors in a case of generalized acquired lipoatrophic diabetes mellitus (Lawrence Syndrome) connected with autoantibodies against adipocyte membranes". ... External links [ edit ] Classification D ICD - 10 : E88.1 External resources Orphanet : 79086 v t e Disorders of subcutaneous fat Panniculitis Lobular without vasculitis Cold Cytophagic histiocytic Factitial Gouty Pancreatic Traumatic needle-shaped clefts Subcutaneous fat necrosis of the newborn Sclerema neonatorum Post-steroid panniculitis Lipodermatosclerosis Weber–Christian disease Lupus erythematosus panniculitis Sclerosing lipogranuloma with vasculitis: Nodular vasculitis / Erythema induratum Septal without vasculitis: Alpha-1 antitrypsin deficiency panniculitis Erythema nodosum Acute Chronic with vasculitis: Superficial thrombophlebitis Lipodystrophy Acquired generalized: Acquired generalized lipodystrophy partial: Acquired partial lipodystrophy Centrifugal abdominal lipodystrophy HIV-associated lipodystrophy Lipoatrophia annularis localized: Localized lipodystrophy Congenital Congenital generalized lipodystrophy Familial partial lipodystrophy Marfanoid–progeroid–lipodystrophy syndrome Poland syndrome
Two conditions in which multiple exostoses occur are metachondromatosis (156250) and the Langer-Giedion syndrome (LGS; 150230); the latter condition is also known as trichorhinophalangeal syndrome type II. Furthermore, exostosis-like lesions occur with fibrodysplasia ossificans progressiva (FOP; 135100), occipital horn syndrome (304150), and the adult stage of hereditary hypophosphatemia (see 307800); these exostoses are located at sites of tendon and muscle attachment. ... They noted that the multiple exostoses of the Langer-Giedion syndrome are indistinguishable from those of the isolated disorder and that at least 1 case of trichorhinophalangeal syndrome without multiple exostoses had been found to have deletion in this region (Hamers et al., 1983). Buhler and Malik (1984) suggested that 2 closely linked loci may be situated at 8q24: 1 for type I trichorhinophalangeal syndrome (TRPS1; 190350) and 1 for multiple exostoses, and both may be deleted in LGS. ... Ludecke et al. (1995) and Hou et al. (1995) presented evidence that the Langer-Giedion syndrome is a true contiguous gene syndrome due to loss of functional copies of both the TRPS1 and the EXT1 gene and that the EXT1 gene is more than 1 Mb distal to the TRPS1 gene.
McGaughran et al. (1995) described a patient with the combination of multiple exostoses and the WAGR syndrome (Wilms tumor, aniridia, genital anomalies, and mental retardation; 194070), a well-documented contiguous gene syndrome resulting from deletion of 11p13. ... As pointed out by Potocki et al. (1995), the description of the contiguous gene syndrome resulting from interstitial deletion of 11p, del(11)(p12p11.2), including multiple exostoses as a feature, provided confirmation of the mapping of EXT2. Other features of this contiguous gene syndrome are mental retardation and parietal foramina, known as Catlin marks (168500).
For a phenotypic description and a discussion of genetic heterogeneity of multiple exostoses, see (133700). Mapping In a child with multiple exostoses with an interstitial deletion of chromosome 11, Le Merrer et al. (1994) excluded linkage to markers in the region 11p12-p11. However, the locus they termed 'EXT2' was mapped to 19p by linkage to a microsatellite DNA marker at the D19S221 locus. In studies of 21 families, they found a maximum location score of 7.22 with a proportion of families (35%) linked to chromosome 19 and a proportion of families (65%) linked to chromosome 8. Their data supported the location of EXT1 distal to D8S198 and the location of the other gene, designated EXT3, between D19S413 and D19S221.
PHACE is an acronym used to describe a syndrome characterised by the association of Posterior fossa brain malformations, large facial Haemangiomas, anatomical anomalies of the cerebral Arteries, aortic coarctation and other Cardiac anomalies, and Eye abnormalities. Sternal anomalies are also sometimes present, and in these cases the syndrome is referred to as PHACES. Two additional manifestations have recently been added to the clinical spectrum of PHACE syndrome: stenosis of the vessels at the base of the skull and segmental longitudinal dilations of the internal carotid artery. Epidemiology This association of manifestations is rare and less than 100 cases have been reported in the literature so far. The syndrome is much more common in females than in males (8:1). ... Expression of the complete clinical picture of PHACE syndrome is extremely rare and most patients display only a partial phenotype (or incomplete clinical spectrum). ... In most cases, the vascular lesions are uni- and homolateral, indicating that PHACE syndrome is a member of the group of cerebrofacial syndromes with involvement of several adjacent segments of the neural crest.
Diagnosis In a consensus statement regarding diagnostic criteria for PHACE syndrome (Metry et al., 2009), the criteria were stratified into 2 categories, PHACE syndrome and possible PHACE syndrome. ... They also reviewed 41 cases with similar findings from the literature, and proposed the use of the acronym PHACE syndrome. Coats et al. (1999) described the ophthalmologic features of PHACE syndrome. ... The patient they reported had congenital glaucoma, a feature not previously reported in association with PHACE syndrome, which required surgical management. ... Oral facial clefting and aortic aneurysms had rarely been reported in PHACE syndrome previously. The aortic aneurysm ruptured after minor trauma at 11 years of age. ... Although the patient had arachnodactyly and some of the other features associated with Marfan syndrome (154700), she did not fulfill the diagnostic criteria.
PHACE syndrome is the association of a large hemangioma , usually on the face or neck, in combination with one or more other birth defects. People with PHACE syndrome may have P osterior fossa brain malformations, H emangioma, A rterial lesions (blood vessel abnormalities in the head or neck), C ardiac (heart) abnormalities/ aortic coarctation , and E ye abnormalities.
Rombo syndrome Other names Vermiculate atrophoderma, milia, hypotrichosis, trichoepitheliomas, basal cell carcinomas and peripheral vasodilation with cyanosis Rombo syndrome is inherited in an autosomal dominant manner [1] Rombo syndrome is a very rare genetic disorder characterized mainly by atrophoderma vermiculatum of the face, [2] : 580 multiple milia , telangiectases , acral erythema , [3] peripheral vasodilation with cyanosis [4] and a propensity to develop basal cell carcinomas . [3] The lesions become visible in late childhood, began at ages 7 to 10 years and are most pronounced on the face, At that time a pronounced, somewhat cyanotic redness of the lips and hands was evident as well as moderate follicular atrophy of the skin on the cheeks. ... The disorder had been transmitted through at least 4 generations with instances of male-to-male transmission. [4] See also [ edit ] Tricho–rhino–phalangeal syndrome List of cutaneous conditions List of cutaneous conditions associated with increased risk of nonmelanoma skin cancer List of cutaneous neoplasms associated with systemic syndromes References [ edit ] ^ "OMIM Entry - 180730 - ROMBO SYNDROME" . omim.org . ... ISBN 0-7216-2921-0 . ^ a b van Steensel MA, Jaspers NG, Steijlen PM (June 2001). "A case of Rombo syndrome". Br. J. Dermatol. 144 (6): 1215–8. doi : 10.1046/j.1365-2133.2001.04235.x . ... S2CID 24195995 . ^ a b c Michaëlsson G, Olsson E, Westermark P (1981). "The Rombo syndrome: a familial disorder with vermiculate atrophoderma, milia, hypotrichosis, trichoepitheliomas, basal cell carcinomas and peripheral vasodilation with cyanosis".
Clinical Features Michaelsson et al. (1981) designated an apparently hitherto undescribed disorder Rombo syndrome after the oldest affected family member. ... The disorder had been transmitted through at least 4 generations with instances of male-to-male transmission. Some similarity to the Bazex syndrome (301845) was noted. Van Steensel et al. (2001) reported a case in which the clinical findings closely matched those described by Michaelsson et al. (1981). ... Van Steensel et al. (2001) noted that the histologic findings are similar to solar elastosis and suggested that the Rombo syndrome gene may be involved in DNA repair and/or cell cycle regulation. ... Inheritance Male-to-male transmission through 4 generations in the family with Rombo syndrome described by Michaelsson et al. (1981) is consistent with autosomal dominant inheritance.
Rombo syndrome is characterized by vermiculate atrophoderma, milia, hypotrichosis, trichoepitheliomas, peripheral vasodilation with cyanosis and basal cell carcinomas.
Harris platelet syndrome Specialty Hematology Harris platelet syndrome (HPS) is the most common inherited giant platelet disorder . [1] [2] Contents 1 Presentation 2 Diagnosis 3 Treatment 4 Prevalence 5 Terminology 6 References Presentation [ edit ] HPS was identified among healthy blood donors in the north-eastern part of the Indian subcontinent, characterized by absent bleeding symptoms, mild to severe thrombocytopenia (platelets rarely <50 X 109/L)with giant platelets (Mean platelet volume 10fL) and normal platelet aggregation studies with absent MYH9 mutation. [1] [2] In the blood donors with HPS authors found a statistically higher MPV, RDW and a lower platelet count and platelet biomass. [3] Diagnosis [ edit ] At present the diagnosis of HPS is made by ascertaining the ethnicity of the patient, as well as assessing for conditions causing acquired thrombocytopenias, and after also excluding the known inherited giant platelet disorders(IGPD) and other congenital thrombocytopenias. ... You can help by adding to it . ( October 2017 ) Prevalence [ edit ] It is extremely important to recognize Harris platelet syndrome, as one third the population of certain parts of Indian subcontinent is affected. [5] Terminology [ edit ] In 2002, this syndrome was called "asymptomatic constitutional macro thrombocytopenia" (ACMT). [1] In 2005, to avoid confusion between ACMT and congenital amegakaryocytic thrombocytopenia (CAMT) this CAMT entity was referred as Harris platelet syndrome . [2] References [ edit ] ^ a b c Naina HV, Nair SC, Daniel D, George B, Chandy M (June 2002). ... PMID 12079722 . ^ a b c Naina HV, Nair SC, Harris S, Woodfield G, Rees MI (November 2005). "Harris syndrome - a geographic perspective". J. ... "Platelet and red blood cell indices in Harris platelet syndrome". Platelets . 21 (4): 303–6. doi : 10.3109/09537101003615402 . ... PMID 16916536 . ^ Naina HV, Harris S (October 2008). "Harris platelet syndrome--underdiagnosed and unrecognized" .
"New World syndrome in Western Australian aborigines" . ... Archived from the original on 2013-01-05. ^ Shell, Ellen Ruppel (2001). "New World Syndrome - Spam and turkey tails have turned Micronesians into Macronesians. ... External links [ edit ] Ellen Ruppel Shell (2001-06-01). "New World Syndrome" . The Atlantic . Ken Weiss (2014-01-08). "The "New World Syndrome" genetic basis: has it been found?" ... The Mermaid's Tale . Ken Weiss (1984). "A new world syndrome of metabolic diseases with a genetic and evolutionary basis".
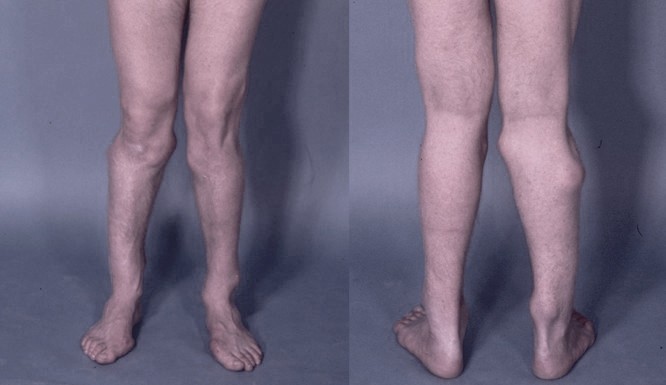
Cantú syndrome is a rare condition characterized by excess hair growth (hypertrichosis), a distinctive facial appearance, heart defects, and several other abnormalities. The features of the disorder vary among affected individuals. People with Cantú syndrome have thick scalp hair that extends onto the forehead and grows down onto the cheeks in front of the ears. ... Most have mildly delayed speech, and some affected children have mild intellectual disability or learning problems. Frequency Cantú syndrome is a rare condition. About three dozen affected individuals have been reported in the medical literature. Causes Cantú syndrome results from mutations in the ABCC9 gene. ... It is unknown how this problem with potassium channel function leads to excess hair growth, heart defects, and the other features of Cantú syndrome. Learn more about the gene associated with Cantú syndrome ABCC9 Inheritance Pattern This condition is inherited in an autosomal dominant pattern, which means one copy of the altered ABCC9 gene in each cell is sufficient to cause the disorder.
Robertson et al. (1999) reported 2 unrelated patients with Cantu syndrome (hypertrichosis, osteochondrodysplasia, and cardiomegaly). ... She also had unusual, deep plantar creases, not previously reported in Cantu syndrome. Robertson et al. (1999) reviewed 14 published cases of Cantu syndrome, including their own. ... Grange et al. (2006) reported a mother and 2 daughters with Cantu syndrome, consistent with autosomal dominant inheritance. ... Cytogenetics Tan et al. (2005) reported a 16-year-old boy with features of Cantu syndrome who was found to have a distal 1p36 deletion (see monosomy 1p36 syndrome, 607872). ... Tan et al. (2005) suggested that patients with Cantu syndrome with unusual or more severe features be analyzed for subtelomeric deletions.
This second gene is also located on the short arm of chromosome 12 (12p12.1). [ citation needed ] Mechanism [ edit ] In terms of the mechanism of Cantú syndrome, mutations in the ABCC9 gene total 25/31. ... "Clinical utility gene card for: Cantú syndrome" . European Journal of Human Genetics . 25 (4): 512. doi : 10.1038/ejhg.2016.185 . ... (August 2002). "Further case of Cantú syndrome: exclusion of cryptic subtelomeric chromosome aberrations". ... PMID 12210352 . ^ Reference, Genetics Home. "Cantú syndrome" . Genetics Home Reference . Retrieved 2017-03-23 . ^ Pubchem. ... "KATP channels and cardiovascular disease: Suddenly a syndrome" . Circulation Research . 112 (7): 1059–1072. doi : 10.1161/CIRCRESAHA.112.300514 .
Each child of an individual with Cantú syndrome has a 50% chance of inheriting the pathogenic variant and being affected. ... Woman age 40 years with Cantù syndrome A. Chest x-ray showing marked cardiomegaly Figure 4. ... Prevalence The prevalence of Cantú syndrome is unknown. To date, about 150 individuals have been reported with Cantú syndrome. Two previously reported conditions, acromegaloid facial appearance (AFA) syndrome and hypertrichosis with acromegaloid facial features (HAFF) syndrome, are now realized to be cases of Cantú syndrome with variable and sometimes milder phenotypic features. Cantú syndrome has been reported worldwide and in all ethnic groups.