-
Hemoperitoneum
Wikipedia
References [ edit ] External links [ edit ] Classification D ICD - 10 : K66.1 ICD - 9-CM : 568.81 MeSH : D006465 v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum v t e Disorders of bleeding and clotting Coagulation · coagulopathy · Bleeding diathesis Clotting By cause Clotting factors Antithrombin III deficiency Protein C deficiency Activated protein C resistance Protein S deficiency Factor V Leiden Prothrombin G20210A Platelets Sticky platelet syndrome Thrombocytosis Essential thrombocythemia DIC Purpura fulminans Antiphospholipid syndrome Clots Thrombophilia Thrombus Thrombosis Virchow's triad Trousseau sign of malignancy By site Deep vein thrombosis Bancroft's sign Homans sign Lisker's sign Louvel's sign Lowenberg's sign Peabody's sign Pratt's sign Rose's sign Pulmonary embolism Renal vein thrombosis Bleeding By cause Thrombocytopenia Thrombocytopenic purpura : ITP Evans syndrome TM TTP Upshaw–Schulman syndrome Heparin-induced thrombocytopenia May–Hegglin anomaly Platelet function adhesion Bernard–Soulier syndrome aggregation Glanzmann's thrombasthenia platelet storage pool deficiency Hermansky–Pudlak syndrome Gray platelet syndrome Clotting factor Hemophilia A/VIII B/IX C/XI von Willebrand disease Hypoprothrombinemia/II Factor VII deficiency Factor X deficiency Factor XII deficiency Factor XIII deficiency Dysfibrinogenemia Congenital afibrinogenemia Signs and symptoms Bleeding Bruise Hematoma Petechia Purpura Nonthrombocytopenic purpura By site head Epistaxis Hemoptysis Intracranial hemorrhage Hyphema Subconjunctival hemorrhage torso Hemothorax Hemopericardium Pulmonary hematoma abdomen Gastrointestinal bleeding Hemobilia Hemoperitoneum Hematocele Hematosalpinx joint Hemarthrosis
-
Multisystem Developmental Disorder
Wikipedia
PMID 12244552 . ^ Turnpenny, PD; Ellard, S (2011). "Alagille syndrome: Pathogenesis, diagnosis and management" . ... PMID 21934706 . ^ Hendrix, JD, Jr; Greer, KE (1996). "Rubinstein-Taybi syndrome with multiple flamboyant keloids". ... PMID 17009066 . ^ Bellugi, U; Wang, PP; Jernigan, TL (1994). "Williams Syndrome: An Unusual Neuropsychological Profile". ... ISBN 978-0-8058-1180-3 . ^ Morris, CA; Lenhoff, HM; Wang, PP (2006-02-13). Williams-Beuren Syndrome: Research, Evaluation, and Treatment . p. 237. ... "Imaging manifestations in Proteus syndrome: An unusual multisystemdevelopmental disorder" .
-
Camptodactyly-Fibrous Tissue Hyperplasia-Skeletal Dysplasia Syndrome
Orphanet
Camptodactyly - fibrous tissue hyperplasia - skeletal dysplasia syndrome is an extremely rare chondrodysplastic malformation syndrome that is characterized by the combination of arachnodactyly, becoming evident at around the age of 10, camptodactyly (hammertoes) and scoliosis. ... Camptodactyly - fibrous tissue hyperplasia - skeletal dysplasia syndrome has been described once in 3 siblings and is suspected to follow autosomal recessive transmission.
-
Trichohepatoenteric Syndrome
Gard
Trichohepatoenteric syndrome is a condition that affects the hair, liver, and intestines. ... Continued diarrhea can lead to an inability to gain weight ( failure to thrive ). Trichohepatoenteric syndrome also causes liver disease such as cirrhosis (chronic liver damage) or hepatomegaly (swollen liver). Affected individuals may have wooly or patchy hair. Tirchohepatoenteric syndrome is caused by mutations in TTC37 or SKIV2L and is inherited in an autosomal recessive manner.TTC37, SKIV2L, HSD11B2, FMR1, AVP, TP53, THRB, PTCH1, PTH, RET, MEN1, MECP2, PTHLH, AR, IGF1, MRC1, NLRP3, VEGFA, IL6, LOXL1, ADIPOQ, FH, LPL, GH1, FRAXA, EHMT1, CYP17A1, KCNQ1, DICER1, INSR, MSH6, NSD1, MSH2, SHANK3, POMC, PMS2, RNU1-4, SCN5A, SNORD116@, TBCE, AMH, VDR, TNF, VHL, SLC26A4, TBX1, GATA2, FOXP3, PDLIM5, SOX10, GLI3, POLR3A, IFNG, TRIM21, RAI1, FBN1, TKT, PAX2, KCNJ2, TXNRD2, CXCR4, REN, ACTG2, LEP, ZEB2, LMX1B, TWIST1, UBE3A, MC2R, VWF, ABCB4, TP63, EP300, KMT2D, TRPV6, FMR1-IT1, BUB1B, NUFIP2, ATRX, CYP19A1, ATM, BBS2, EDA, AQP4, ACE, HPSE2, EGF, FOXL2, CRP, APC, ELN, ADRB1, B3GLCT, CREBBP, FLCN, ZFP36, SYCE1, NSD2, BTBD9, USH1G, MKRN3, WFS1, PROKR2, WNT7A, ZNF217, NPHS2, CNBP, TTC7A, ALMS1, TUBA1A, BUD13, MAEL, SUCLA2, SLC4A4, TNKS, DISP1, PLA2G6, LOH19CR1, SMC1A, MKKS, MIA, HMGA2, IL33, FGF23, CNTN4, TMEM67, SLC35A2, VPS13B, CBLL2, KANSL1, NKX2-1, GOLGA6A, MIR93, THPO, TH, TFRC, TFPI, TECTA, TCOF1, TCN2, TBX5, MIR597, PTLS, PGR-AS1, TNFRSF1B, TNXB, TRH, ASXL1, UVRAG, KDM6A, CLRN1, UROD, SGCE, ADAMTS18, RLS1, RFX6, TTR, UBR1, TTF1, TSPY1, TSHR, TRPS1, ST3GAL5, LGI1, SMC3, FTSJ1, MLH3, GBA2, CNNM4, ZBTB20, SETBP1, NIPBL, LMOD1, MARK4, LGALS13, CCNDBP1, ZNF462, ADNP, SATB2, ABRAXAS2, SETX, PUF60, FOXP1, IL22, SLC33A1, DARS2, POLR3B, NLRP2, CHD7, FERMT1, SHROOM4, CHDH, RCBTB1, AHI1, ARHGEF3, UGT1A1, LARP7, IL23A, WDR35, CRBN, TSHZ3, F11R, B4GALT7, TREX1, SST, LPIN2, MAFB, CDC73, MUL1, KMT2B, USB1, CEP290, COL18A1, GOSR2, CPAT1, BAG3, CHST3, EIF2AK3, FIP1L1, ATF7IP, EPPK1, MFRP, ABCB6, HUWE1, PQBP1, PLEKHF1, SPINK5, PROK2, SMOC1, KCNQ1OT1, CKAP4, MORF4, XYLT2, CTCF, IFIH1, STRA6, PORCN, AGPAT2, SLC9A6, TUBA1B, ZMPSTE24, STK11, ACTB, RO60, FGFR2, FGF3, FGD1, ACSL3, FANCC, F9, F2, EYA1, ERCC6, ERCC3, EPHX1, ENG, EIF2S3, EEC1, EDNRA, TOR1A, DRD3, DRD2, DNMT3B, DLX6, FGFR3, FOXC1, HMGN1, FLT1, NR3C1, GRIK2, GPT, GRK5, GNAS, GLI1, GPC3, GJA1, CBLIF, GHRHR, GHR, GEM, GBA, GATA3, GATA1, GALT, GABRG2, FTL, FSHB, DLX5, DLD, DIO3, DHCR7, CD34, CD6, CBR1, CALR, CAD, BRCA2, BRCA1, BDNF, BCL2, BCKDHA, AVPR2, ATR, STS, FAS, APOE, SLC25A4, ANPEP, AHSG, AGTR1, CD59, CDC42, CDK4, CTNNB1, DGCR, TIMM8A, DCX, CYP21A2, CYP11B2, CYP11B1, CYP11A1, CYP2B6, SLC25A10, CHGA, CRKL, COL4A5, COL3A1, COL2A1, COL1A2, COL1A1, LYST, CHRM1, HLA-DRB5, HMOX2, SREBF1, PTGS2, PRNP, MAPK1, PTPA, PPARG, POR, PON1, POLG, POLA1, PKP1, PITX2, PIK3CA, PIGA, PGF, PDE3A, PRKN, CNTN3, SERPINE1, PAFAH1B1, OTC, PTEN, PTX3, HPGD, RAD51, SOX2, SOD1, SMARCE1, SMARCB1, SLC16A2, SLC5A2, SKI, SDHD, CXCL12, CCL11, SCN4A, SCN1A, RYR2, RYR1, RPS19, RPS6, RPE65, RBBP8, RASA1, TNFRSF11B, OCRL, NTRK1, NOTCH3, LIG1, LIF, KRT8, KIR3DL1, KDR, KCNQ2, KCNJ1, KCNH2, INSL4, INS, CXCL10, IL17A, IL4, IL2, IL1B, IL1A, IGF2, IDS, HTC2, LRP5, SH2D1A, EPCAM, ATP8, NOS3, NDUFAB1, NBN, MYO5B, MYLK, MYH11, MYH9, MUTYH, ATP6, MAG, MMP2, NR3C2, MLH1, MEST, MC4R, MAS1, MAP6, MAOA, PEX2

-
Multinodular Goiter-Cystic Kidney-Polydactyly Syndrome
Orphanet
Multinodular goiter - cystic kidney - polydactyly syndrome is a very rare syndrome characterized by the association of multinodular goiter, cystic renal disease and digital anomalies. ... Goiter and/or digitalized thumbs and/or polydactyly were present in other members of families. Genetic counseling This syndrome seems to be transmitted as an autosomal dominant trait with variable expression and incomplete penetrance.
-
Tel Hashomer Camptodactyly Syndrome
Orphanet
Tel Hashomer camptodactyly syndrome is a rare syndrome characterized by camptodactyly, muscle hypoplasia and weakness, skeletal anomalies, facial dysmorphism and abnormal dermatoglyphics. ... Etiology The molecular basis of the syndrome has not yet been elucidated. Genetic counseling Inheritance is probably autosomal recessive.
-
Intractable Diarrhea-Choanal Atresia-Eye Anomalies Syndrome
Orphanet
Intractable diarrhea-choanal atresia-eye anomalies syndrome is characterised by the association of intractable diarrhoea of infancy with choanal atresia. Short stature, a prominent and broad nasal bridge, micrognathia, single palmar creases, chronic corneal inflammation, cytopenia, and abnormal hair texture were also reported. So far, the syndrome has been described in three children from the same family. The absence of intellectual deficit and immune deficiency allow this syndrome to be distinguished from other forms of intractable diarrhoea of infancy described previously.
-
Insulin Autoimmune Syndrome
Gard
Insulin autoimmune syndrome is a rare condition that causes low blood sugar ( hypoglycemia ). ... People affected by insulin autoimmune syndrome have antibodies that attack insulin, causing it to work too hard and the level of blood sugar to become too low. Insulin autoimmune syndrome most often begins during adulthood.
-
Hepatorenal Syndrome
Gard
Hepatorenal syndrome is a form of impaired kidney function that occurs in individuals with advanced chronic liver disease. As many as 40% of individuals with cirrhosis and ascites will develop hepatorenal syndrome. Symptoms may include fatigue, abdominal pain, and a general feeling of ill health ( malaise ). There are two distinct types of hepatorenal syndrome. Type I progresses quickly (within days), leading to kidney failure . ... Type II progresses more slowly, over weeks or months, and the symptoms are less severe. The cause of hepatorenal syndrome is unknown. A contributing factor seems to be a narrowing of the blood vessels that connect into the kidneys.HMOX1, EDN1, TNF, TLR4, ALB, RSC1A1, HARS1, ATN1, HGS, SRSF5, LCN2, CST3, VCAM1, RAPGEF5, NR1I2, MIR21, MIR146A, ADM, TYR, TIMP2, ITPR1, CXCL10, IGFBP7, CSF3, MIR210
-
Corneodermatoosseous Syndrome
Wikipedia
Corneodermatoosseous syndrom Other names CDO syndrome [1] This condition is inherited in an autosomal dominant manner Corneodermatosseous syndrome is an autosomal dominant condition with onset in infancy, characterized by corneal dystrophy, photophobia, diffuse palmoplantar keratoderma , distal onycholysis, skeletal abnormalities, with brachydactyly, short stature, and medullary narrowing of digits. [2] See also [ edit ] Palmoplantar keratoderma Keratoderma Skin lesion Terminal osseous dysplasia with pigmentary defects List of cutaneous conditions References [ edit ] ^ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Corneodermatoosseous syndrome" . www.orpha.net . Retrieved 19 April 2019 . ^ Stevens, Howard P.; David P.

-
Rh Deficiency Syndrome
Gard
The Rh deficiency syndrome , also known as Rh-null syndrome, is a blood disorder where people have red blood cells (RBCs) lacking all Rh antigens . ... There are two types of Rh deficiency syndrome: The regulator type is associated with many different changes (mutations) in the RHAG gene . ... These patients are at risk of having adverse transfusion reactions because they may produce antibodies against several of the Rh antigens and can only receive blood from people who have the same condition. Rh deficiency syndrome is inherited in an autosomal recessive manner.
-
Cyprus Facial-Neuromusculoskeletal Syndrome
Orphanet
Cyprus facial-neuromusculoskeletal syndrome is an exceedingly rare, genetic malformation syndrome characterized by a striking facial appearance, variable skeletal deformities, and neurological defects. Epidemiology The syndrome has been described in a single Greek Cypriot family, over three generations. ... Other manifestations include restricted joint stiffness, ankyloses, ptosis, and cataracts. Etiology The cause of this syndrome is not known. Genetic counseling This condition is likely to be autosomal dominant.
-
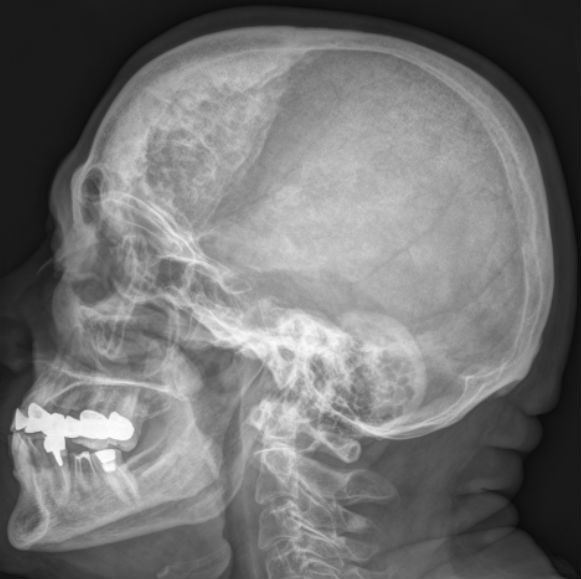
Katz Syndrome
Wikipedia
Katz syndrome Other names Hyperostosis frontalis interna Hyperostosis frontalis interna in a 74-year-old woman Specialty Medical genetics Katz syndrome is a rare congenital disorder , presenting as a polymalformative syndrome characterized by enlarged viscera , hepatomegaly , diabetes , and skeletal anomalies that result in a short stature, cranial hyperostosis , and typical facial features. ... You can help by adding to it . ( September 2017 ) References [ edit ] ^ Bruno Bissonnette, Igor Luginbuehl, Bruno Marciniak, Bernard J. Dalens (eds.): Syndromes: Rapid Recognition and Perioperative Implications (McGraw-Hill Companies, 2006) ISBN 0-07-135455-7 This article about a congenital malformation is a stub .
-
Malouf Syndrome
Wikipedia
Malouf syndrome Other names Dilated cardiomyopathy-hypergonadotropic hypogonadism syndrome Malouf syndrome (also known as "congestive cardiomyopathy-hypergonadotropic hypogonadism syndrome") is a congenital disorder that causes one or more of the following symptoms: mental retardation , ovarian dysgenesis , congestive cardiomyopathy , broad nasal base, blepharoptosis , and bone abnormalities, and occasionally marfanoid habitus (tall stature with long and thin limbs, little subcutaneous fat , arachnodactyly , joint hyperextension , narrow face, small chin, large testes, and hypotonia ). [2] This disease is named after J.
-
Deafness And Myopia Syndrome
Medlineplus
Deafness and myopia syndrome is a disorder that causes problems with both hearing and vision. ... Frequency The prevalence of deafness and myopia syndrome is unknown. Only a few affected families have been described in the medical literature. Causes Deafness and myopia syndrome is caused by mutations in the SLITRK6 gene. ... SLITRK6 gene mutations that cause deafness and myopia syndrome result in an abnormally short SLITRK6 protein that is not anchored properly to the cell membrane. ... Impaired SLITRK6 protein function leads to abnormal nerve development in the inner ear and improperly controlled eyeball growth, resulting in the hearing loss and nearsightedness that occur in deafness and myopia syndrome. Learn more about the gene associated with Deafness and myopia syndrome SLITRK6 Inheritance Pattern This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have mutations.
-
Complement 4 Deficiency
Wikipedia
External links [ edit ] Classification D OMIM : 120820 DiseasesDB : 1873 v t e Lymphoid and complement disorders causing immunodeficiency Primary Antibody / humoral ( B ) Hypogammaglobulinemia X-linked agammaglobulinemia Transient hypogammaglobulinemia of infancy Dysgammaglobulinemia IgA deficiency IgG deficiency IgM deficiency Hyper IgM syndrome ( 1 2 3 4 5 ) Wiskott–Aldrich syndrome Hyper-IgE syndrome Other Common variable immunodeficiency ICF syndrome T cell deficiency ( T ) thymic hypoplasia : hypoparathyroid ( Di George's syndrome ) euparathyroid ( Nezelof syndrome Ataxia–telangiectasia ) peripheral: Purine nucleoside phosphorylase deficiency Hyper IgM syndrome ( 1 ) Severe combined (B+T) x-linked: X-SCID autosomal: Adenosine deaminase deficiency Omenn syndrome ZAP70 deficiency Bare lymphocyte syndrome Acquired HIV/AIDS Leukopenia : Lymphocytopenia Idiopathic CD4+ lymphocytopenia Complement deficiency C1-inhibitor ( Angioedema / Hereditary angioedema ) Complement 2 deficiency / Complement 4 deficiency MBL deficiency Properdin deficiency Complement 3 deficiency Terminal complement pathway deficiency Paroxysmal nocturnal hemoglobinuria Complement receptor deficiency This immunology article is a stub .
-
Grisel's Syndrome
Wikipedia
Grisel's syndrome Specialty Rheumatology Grisel’s syndrome is a non-traumatic subluxation of the atlanto-axial joint caused by inflammation of the adjacent tissues. ... Post-operative inflammation after certain procedures such as adenoidectomy can also lead to this condition in susceptible individuals such as those with Down syndrome . Contents 1 Pathophysiology 2 Diagnosis 3 Treatment 4 References 5 External links Pathophysiology [ edit ] Pathophysiology of this disease consists of relaxation of the transverse ligament of the atlanto-axial joint. Diagnosis [ edit ] Diagnosis can be established using plain film x-rays as well as CT scan of the neck/cervical spine. Children with Down syndrome have inherently lax ligaments making them susceptible to this condition. ... Mathern GW, Batzdorf U. Grisel's syndrome: Cervical spine clinical, pathologic, and neurologic manifestations . ... C Bocciolini, D Dall’Olio, E Cunsolo, PP Cavazzuti, and P Laudadio, Grisel’s syndrome: a rare complication following adenoidectomy , Acta Otorhinolaryngol Ital. 2005 August; 25(4): 245–249.
-
Stratton Parker Syndrome
Wikipedia
Stratton-Parker Syndrome Other names Short stature wormian bones dextrocardia Stratton parker syndrome is a rare disorder characterized by short stature, wormian bones (extra cranial bones), and dextrocardia (displaced heart). [1] Other symptoms include dermatoglyphics , tooth deformities or missing teeth, abnormal kidney development, shortened limbs, mental retardation, undescended testes or cryptorchidism , and anal atresia . [1] The condition was first described by Stratton and Parker in 1989, [2] and there have been only four reported cases worldwide. [3] Two cases of the syndrome were reported by Gilles-Eric Seralini in 2010 after having been contacted in January 2009. [4] Alternative names include "Growth Hormone Deficiency with Wormian Bones, Cardiac Anomaly, and Brachycamptodactyly" [5] and "Short stature wormian bones dextrocardia" [1] References [ edit ] ^ a b c "Short stature wormian bones dextrocardia" . ... "Growth hormone deficiency and complex congenital abnormalities: a further case of Stratton-Parker syndrome?" . Endocrine Abstracts. P86 . ... "Two cases of birth defects overlapping Stratton-Parker syndrome after multiple pesticide exposure". ... PMID 19951932 . ^ "STRATTON-PARKER SYNDROME" . BioGraph . Retrieved 2011-09-27 .
-
Oculoauricular Syndrome
Wikipedia
Oculoauricular syndrome Oculoauricular syndrome is inherited in an autosomal recessive manner. Specialty Medical genetics Oculoauricular syndrome is a rare genetic condition affecting the eyes and ears. ... It is also known as the Schorderet-Munier-Franceschetti syndrome. Contents 1 Signs and symptoms 1.1 Eyes 1.2 Ears 2 Genetics 3 Pathogensis 4 Diagnosis 4.1 Differential diagnosis 5 Epidemiology 6 History 7 References 8 External links Signs and symptoms [ edit ] The clinical features of this condition are as follows: [ citation needed ] Eyes [ edit ] microphthalmia coloboma nystagmus corneal sclerosis cataract glaucoma anterior synechiae posterior synechiae macular hypoplasia rod-cone dystrophy divergent strabismus posterior embryotoxon Ears [ edit ] malformed pinnae low-set pinnae crumpled helix narrow external acoustic meatus coloboma of the lobules Hearing is normal Genetics [ edit ] This condition is inherited in an autosomal recessive manner. The gene responsible is located on the short arm of chromosome 4 (4p16.1) [1] Pathogensis [ edit ] This is not presently understood. [ citation needed ] Diagnosis [ edit ] Differential diagnosis [ edit ] This includes Morning glory syndrome [ citation needed ] Epidemiology [ edit ] This condition has only been described in three families to date (2017). [ citation needed ] History [ edit ] This condition was first described in 1945. [2] The gene responsible was identified in 2008. [1] References [ edit ] ^ a b Schorderet, D.F.; Nichini, O.; Boisset, G.; Polok, B.; Tiab, L.; Mayeur, H.; Raji, B; de la Houssaye, G; Abitbol, M.M.; Munier, FL (2008). "Mutation in the human homeobox gene NKX5-3 causes an oculo-auricular syndrome" . American Journal of Human Genetics . 82 (5): 1178–1184. doi : 10.1016/j.ajhg.2008.03.007 .

-
Serkal Syndrome
Wikipedia
Serkal syndrome Other names SEx Reversion, Kidneys, Adrenal and Lung dysgenesis SERKAL syndrome ( SE x R eversion, K idneys, A drenal and L ung dysgenesis) is an autosomal recessive disorder in XX humans. ... The main outcome is female to male sex reversal. [1] Other names include sex reversion-kidneys and adrenal and lung dysgenesis syndrome. The condition's prevalence is lower than 1 in 1,000,000. [2] Contents 1 Presentation 2 Genetics 3 Diagnosis 4 References 5 External links Presentation [ edit ] The effect of the disorder is female to male sex reversal. ... U.; Okopnik, M.; Knopf, C.; Indelman, M.; Drugan, A.; Tiosano, D.; Gershoni-Baruch, R.; Choder, M.; Sprecher, E. (2008). "SERKAL Syndrome: An Autosomal-Recessive Disorder Caused by a Loss-of-Function Mutation in WNT4" . ... PMID 18179883 . ^ "Orphanet: SERKAL syndrome" . www.orpha.net . Retrieved 2019-03-01 . External links [ edit ] SERKAL syndrome ( The Monarch Initiative ) This article about a disease , disorder, or medical condition is a stub .




