Lewis-Sumner syndrome (also known as multifocal acquired demyelinating sensory and motor neuropathy) is a neurological condition affecting primarily the arms and hands (upper limbs). The symptoms are a result of inflammation of the nerves leading to the upper body and the destruction of the fatty covering that protects the nerves ( myelin sheath ). Lewis Sumner syndrome is an acquired disorder, and the exact cause of the condition is not known. Lewis Sumner syndrome may be difficult to distinguish from other forms of demyelinating neuropathies , but diagnosis may be possible through nerve conduction studies or a specific type of imaging test, called MRI with T2 STIR .
Lewis-Sumner syndrome (LSS) is a rare acquired demyelinating polyneuropathy characterized by asymmetrical distal weakness of the upper or lower extremities and motor dysfunction with adult onset.
The mitochondrial DNA (mtDNA) depletion syndrome (MDS) is a clinically heterogeneous group of mitochondrial disorders characterized by a reduction of the mtDNA copy number in affected tissues without mutations or rearrangements in the mtDNA. ... Additional phenotypes include fatal infantile lactic acidosis with methylmalonic aciduria, spastic ataxia (early-onset spastic ataxia-neuropathy syndrome), and Alpers syndrome.
For a similarly abbreviated (MdDS) rare neurological condition, see Mal de debarquement . Mitochondrial DNA depletion syndrome Other names mtDNA depletion syndrome Mitochondrial DNA depletion syndrome is inherited via autosomal recessive manner Mitochondrial DNA depletion syndrome ( MDS or MDDS ) is any of a group of autosomal recessive disorders that cause a significant drop in mitochondrial DNA in affected tissues. ... PMID 24741716 . ^ This form of MDDS is also called "Alpers' disease", also called "Alpers' syndrome", "Alpers-Huttenlocher syndrome", "progressive sclerosing poliodystrophy", and "progressive infantile poliodystrophy". ... "Molecular insight into mitochondrial DNA depletion syndrome in two patients with novel mutations in the deoxyguanosine kinase and thymidine kinase 2 genes". ... "SUCLA2-Related Mitochondrial DNA Depletion Syndrome, Encephalomyopathic Form, with Mild Methylmalonic Acuduria" . ... "MPV17-associated hepatocerebral mitochondrial DNA depletion syndrome: new patients and novel mutations".
A number sign (#) is used with this entry because mal de Meleda is caused by homozygous mutation in the SLURP1 gene (606119) on chromosome 8q24. Clinical Features Congenital symmetrical cornification of the palms and soles, with ichthyotic changes elsewhere, characterizes this disorder which derives its name from its relatively high frequency among inhabitants of the Island of Meleda, Dalmatia, Yugoslavia. Bosnjakovic (1938) studied the family in Mljet (or Meleda). Schnyder et al. (1969) provided more recent observations. Hyperhidrosis, perioral erythema, and lichenoid plaques were also noted. Franceschetti et al. (1972) reported on Meleda disease and stated that Neumann (1898) was the first to report the disorder, in 5 families from Meleda.
Mal de Meleda (MdM) is a diffuse palmoplantar keratoderma, initially reported in the Island of Meleda, characterized by symmetric palmoplantar hyperkeratosis that progressively extends to the dorsal surfaces of hands and feet (transgrediens). The disease can be associated to hyperhidrosis, lichenoid plaques and perioral erythema.
Mal de Meleda is a rare skin disorder that begins in early infancy. Affected individuals have a condition known as palmoplantar keratoderma, in which the skin of the palms of the hands and soles of the feet becomes thick, hard, and callused. In mal de Meleda, the thickened skin is also found on the back of the hands and feet and on the wrists and ankles. In addition, affected individuals may have rough, thick pads on the joints of the fingers and toes and on the elbows and knees. Some people with mal de Meleda have recurrent fungal infections in the thickened skin, which can lead to a strong odor. Other features of this disorder can include short fingers and toes (brachydactyly), nail abnormalities, red skin around the mouth, and excessive sweating (hyperhidrosis).
Facial anomalies are not specific for the syndrome, and clinical expression appears to be variable. ... Etiology The genetic mechanism underlying ACFS is still unknown. Isolated or syndromic SHFM has been linked to different loci or genes. ... However, EEC syndrome, Rapp-Hodgkin syndrome (see these terms) and ectrodactyly-cleft lip/palate-hand/foot deformities-intellectual deficit can be ruled out based on lack of ectodermal involvement. Malpuech syndrome (see this term) can also be excluded based on distinct facial features and absent limb defects. CHD, cleft palate, and genital anomalies are features of genito-palato-cardiac syndrome, but none of the reported cases had ectrodactyly.
Guion-Almeida et al. (2000) proposed the name acrocardiofacial syndrome for this condition. Mingarelli et al. (2005) reported an affected male infant who died at age 1 month from cardiorespiratory failure.
Sinding-Larsen and Johansson syndrome Other names Aseptic necrosis of patella The site of the osg and sjs on the knee: OSG at tibial tuberosity and SLJ at inferior pole of patella Sinding-Larsen and Johansson syndrome , [1] named after Swedish surgeon Sven Christian Johansson (1880-1959), [2] and Christian Magnus Falsen Sinding-Larsen (1866-1930), [3] a Norwegian physician, is apophysitis of the inferior pole of the patella . ... This condition called Sinding-Larsen and Johansson syndrome was described independently by Sinding-Larsen in 1921 and Johansson in 1922. [4] Contents 1 Signs and symptoms 2 Diagnosis 3 Treatment 4 References 5 External links Signs and symptoms [ edit ] Patella, its tendon and tibial tuberosity The condition is usually seen in athletic individuals typically between 10–14 years of age. ... If rest fails to provide relief, the abnormal area is removed and the paratenon is stripped. [ citation needed ] References [ edit ] ^ Sinding-Larsen and Johansson syndrome at Who Named It? ^ Sven Christian Johansson at Who Named It? ... ^ APLEYS system of orthopaedics 9th edition External links [ edit ] Classification D ICD - 10 : M92.4 External resources Orphanet : 97337 Wikimedia Commons has media related to Osgood–Schlatter disease . v t e Bone and joint disease Bone Inflammation endocrine : Osteitis fibrosa cystica Brown tumor infection : Osteomyelitis Sequestrum Involucrum Sesamoiditis Brodie abscess Periostitis Vertebral osteomyelitis Metabolic Bone density Osteoporosis Juvenile Osteopenia Osteomalacia Paget's disease of bone Hypophosphatasia Bone resorption Osteolysis Hajdu–Cheney syndrome Ainhum Gorham's disease Other Ischaemia Avascular necrosis Osteonecrosis of the jaw Complex regional pain syndrome Hypertrophic pulmonary osteoarthropathy Nonossifying fibroma Pseudarthrosis Stress fracture Fibrous dysplasia Monostotic Polyostotic Skeletal fluorosis bone cyst Aneurysmal bone cyst Hyperostosis Infantile cortical hyperostosis Osteosclerosis Melorheostosis Pycnodysostosis Joint Chondritis Relapsing polychondritis Other Tietze's syndrome Combined Osteochondritis Osteochondritis dissecans Child leg: hip Legg–Calvé–Perthes syndrome tibia Osgood–Schlatter disease Blount's disease foot Köhler disease Sever's disease spine Scheuermann's_disease arm: wrist Kienböck's disease elbow Panner disease
Sinding-Larsen-Johansson disease is a type of osteochondrosis affecting the attachment of the patellar tendon to the patella and characterised by tenderness and localized swelling of the patella. Epidemiology Prevalence is unknown. Clinical description This disease occurs mostly in adolescents, especially in young male athletes practising sport. Bone fragmentation of the inferior portion of the patella may occur. Etiology It results from contusion or traction tendinitis of the proximal attachment of the patella tendon, followed by de novo calcification. Prognosis The course of the disease is usually self-limited and benign.
Mesomelia-Synostoses syndrome (MSS) is a syndromal osteochondrodysplasia due to a contiguous gene deletion syndrome, characterized by progressive bowing of forearms and forelegs leading to mesomelia, progressive intracarpal or intratarsal bone fusion and fusion of metacarpal bones with proximal phalanges, ptosis, hypertelorism, abnormal soft palate, congenital heart defect, and ureteral anomalies. ... Clinical description In contrast to other mesomelic syndromes, MSS mostly manifests in postnatal life and has a slow progressive clinical course at least until adulthood (when skeletal growth has ceased). ... MSS is likely to represent a contiguous gene deletion syndrome. There is no disorder linked to point mutations of these genes. ... Other rare mesomelic dysplasias, i.e., Langer mesomelic dysplasia or Fryns type micromelic dwarfism (see these terms) are not associated with synostoses. Syndromes with synostoses i.e. Nievergelt syndrome, proximal symphalangism, Osebold-Remondini syndrome and multiple synostoses (see these terms) have different associated anomalies.
A number sign (#) is used with this entry because of evidence that mesomelia-synostoses syndrome is a contiguous gene deletion syndrome caused by heterozygous microdeletion on chromosome 8q13. Description The Verloes-David-Pfeiffer mesomelia-synostoses syndrome is an autosomal dominant form of mesomelic dysplasia comprising typical acral synostoses combined with ptosis, hypertelorism, palatal abnormality, congenital heart disease, and ureteral anomalies (summary by Isidor et al., 2009). ... Isidor et al. (2009) reviewed the clinical features of 5 reported patients with the mesomelia-synostoses syndrome (Verloes and David, 1995; Pfeiffer et al., 1995; Day-Salvatore and McLean, 1998; Leroy et al., 2003) and provided follow-up on 3 of the patients. In contrast to other mesomelic syndromes, the clinical course of this mesomelic dysplasia is slowly progressive, at least until adulthood, with development of severe limb deformities despite repeated corrective surgical intervention. ... Molecular Genetics Using whole-genome oligonucleotide array CGH, Isidor et al. (2010) identified a microdeletion on chromosome 8q13 in each of 5 patients with the mesomelia-synostosis syndrome from 4 previously reported families (Verloes and David, 1995; Pfeiffer et al., 1995; Day-Salvatore and McLean, 1998; and Leroy et al., 2003, respectively).
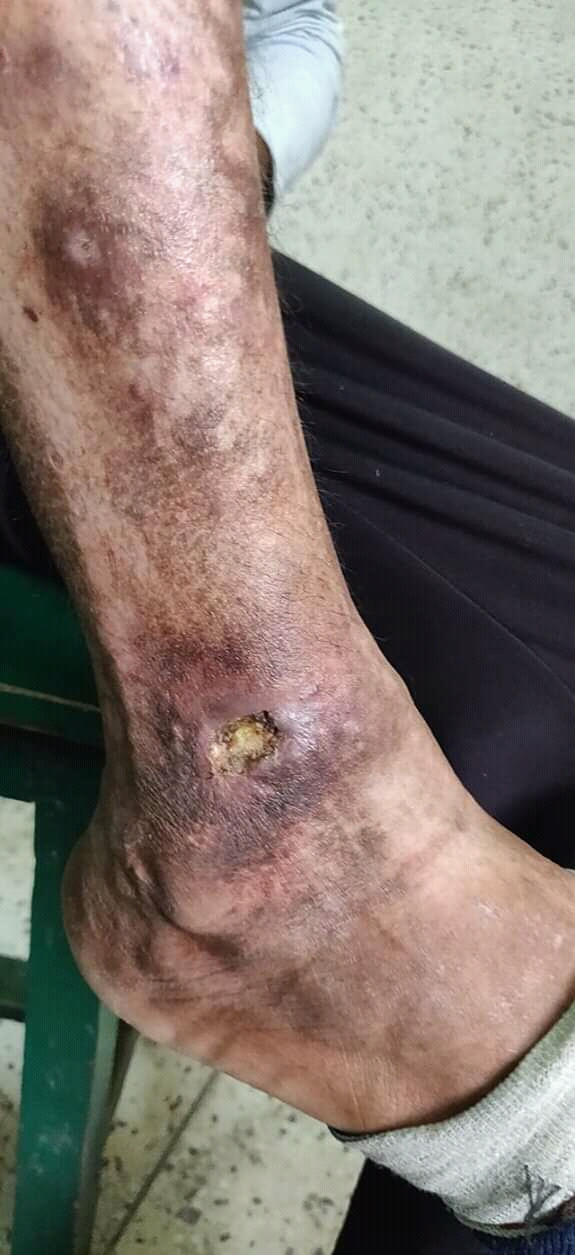
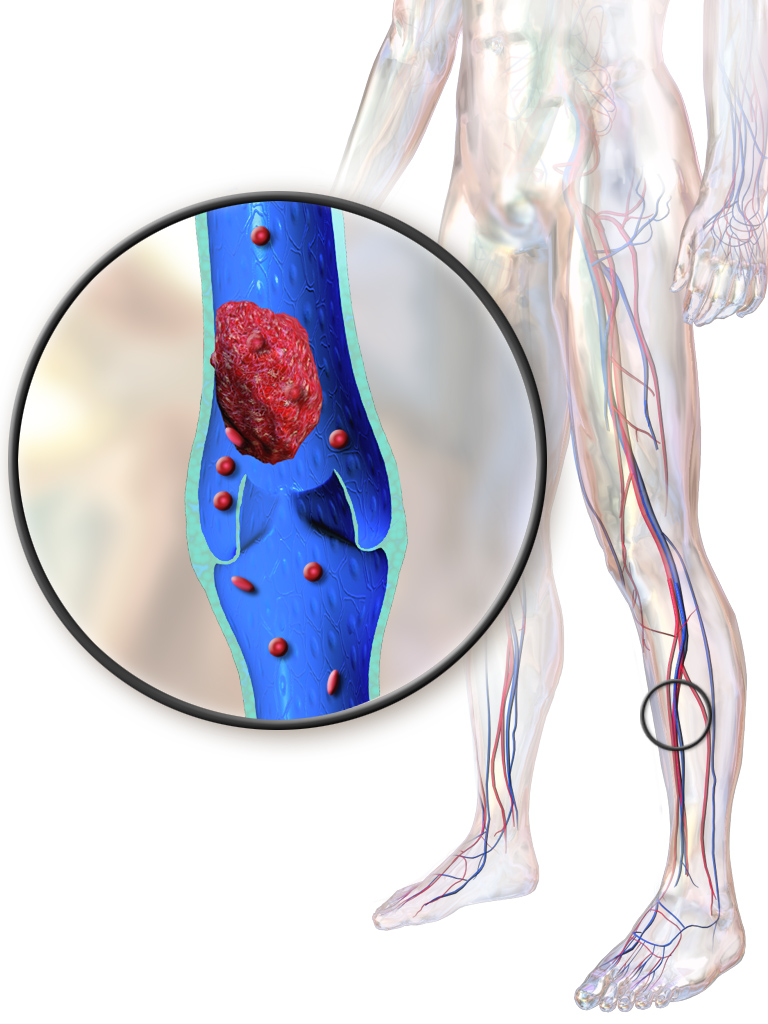
A rare vascular skin disease characterized by recurrent focal non-inflammatory thrombosis of dermal venulae, predominantly of the lower extremities, resulting in a cutaneous response manifested as pruritus and painful papules and erythematous plaques. The lesions evolve into hemorrhagic vesicles or bullae, which rupture and turn into painful ulcers merging into reticulate, confluent, geometric, and painful ulcerations. During a period of a few months, the ulcerations change to porcelain-white atrophic scars with punctate telangiectasia (so-called atrophie blanche). In active disease, lesions in different stages coexist.
Livedoid vasculopathy is a blood vessel disorder that causes painful ulcers and scarring ( atrophie blanche ) on the feet and lower legs. These symptoms can persist for months to years and the ulcers often recur. Livedoid vasculopathy lesions appear as painful red or purple marks and spots that may progress to small, tender, irregular ulcers. Symptoms tend to worsen in the winter and summer months, and affect women more often then men. Livedoid vasculopathy may occur alone or in combination with another condition, such as lupus or thrombophilia.
Hughes–Stovin syndrome Deep vein thrombosis is one of the characteristics of this syndrome Specialty Immunology Hughes–Stovin syndrome is a rare autoimmune disorder of unknown cause that is characterized by the combination of multiple pulmonary artery aneurysms and deep vein thromboses . ... PMID 13618502 . ^ "Hughes stovin syndrome" . Medcyclopaedia . GE . ^ a b c d e f Chalazonitis; et al. (January 29, 2009). "Hughes-Stovin Syndrome: a case report and review of the literature" . ... PMID 19178695 . ^ a b Robinson, C; Miller, D; Will, M; Dhaun, N; Walker, W (2018). "Hughes–Stovin syndrome: the diagnostic and therapeutic challenges of peripheral pulmonary artery aneurysms" . ... PMID 29860510 . ^ a b Khalid, Umair; Saleem, Taimur (2011). "Hughes-Stovin Syndrome" . Orphanet Journal of Rare Diseases . 6 (1): 15. doi : 10.1186/1750-1172-6-15 .
RAB18 deficiency is the molecular deficit underlying both Warburg micro syndrome (characterized by eye, nervous system, and endocrine abnormalities) and Martsolf syndrome (characterized by similar – but milder – findings). ... Liegel et al [2013] Clinical Characteristics Clinical Description RAB18 deficiency describes the molecular deficit underlying Warburg micro syndrome and Martsolf syndrome. Warburg micro syndrome is characterized by eye, nervous system, and endocrine abnormalities [Warburg et al 1993]; Martsolf syndrome is characterized by similar findings, but with a milder presentation [Martsolf et al 1978]. ... Neurologic Findings Intellectual disability (ID). Individuals with Warburg micro syndrome have severe to profound ID. Individuals with Martsolf syndrome have mild to moderate ID. ... Hypogonadism Both Warburg micro syndrome and Martsolf syndrome are frequently associated with hypogonadism. ... Warburg micro syndrome type 1: RAB3GAP1 Warburg micro syndrome type 2: RAB3GAP2 Warburg micro syndrome type 3: RAB18 Warburg micro syndrome type 4: TBC1D20 Prevalence Data on the prevalence of RAB18 deficiency are limited.
PDE4D haploinsufficiency syndrome is a rare syndromic intellectual disability characterized by developmental delay, intellectual disability, low body mass index, long arms, fingers and toes, prominent nose and small chin.
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome on chromosome 5q12. Clinical Features Jaillard et al. (2011) reported 4 unrelated children with developmental delay or mental retardation and facial dysmorphism who were found to have small heterozygous de novo deletions of chromosome 15q12. ... INHERITANCE - Autosomal dominant GROWTH Weight - Decreased body mass index Other - Marfanoid habitus - Growth retardation HEAD & NECK Face - Large forehead - Micrognathia - Small chin - Long philtrum Ears - Posteriorly rotated ears Eyes - Long palpebral fissures - Esotropia - Strabismus - Ptosis Nose - Prominent nose - Prominent columella CARDIOVASCULAR Vascular - Low blood pressure SKELETAL Hands - Long fingers Feet - Long toes MUSCLE, SOFT TISSUES - Hypotonia NEUROLOGIC Central Nervous System - Delayed psychomotor development - Speech delay - Intellectual disability - Seizures (in some patients) Behavioral Psychiatric Manifestations - Autistic features MOLECULAR BASIS - Contiguous gene syndrome involving deletion of 2.63 Mb on chromosome 5q12 ▲ Close
Foreign accent syndrome Specialty Psychiatry , Neurology Foreign accent syndrome is a medical condition in which patients develop speech patterns that are perceived as a foreign accent [1] that is different from their native accent, without having acquired it in the perceived accent's place of origin. ... See also [ edit ] Apraxia of speech Susac's syndrome Xenoglossia References [ edit ] ^ a b c Kurowski, K. ... "Suprasegmental aspects of Foreign Accent Syndrome". In Stojanovik, V.; Setter, J. ... Retrieved 2 September 2019 . ^ "Foreign Accent Syndrome (FAS) Support" . Utdallas.edu . ... London. 14 September 2010. ^ "Coping with Foreign Accent Syndrome" . BBC News . 13 September 2010 .
8p23.1 duplication syndrome Other names Dup(8)(p23.1p23.1), Trisomy 8p23.1 Chromosome 8 is associated with this condition 8p23.1 duplication syndrome is a rare genetic disorder caused by a duplication of a region from human chromosome 8 . [1] This duplication syndrome has an estimated prevalence of 1 in 64,000 births [1] and is the reciprocal of the 8p23.1 deletion syndrome . ... Duplication of the GATA4 transcription factor (OMIM: 600576 ) is believed to underlie the congenital heart disease and other genes, common to the duplication and deletion syndromes, can be regarded as candidates for the 8p23.1 duplication syndrome. ... The diaphragmatic hernia found in the 8p23.1 deletion syndrome has not been found in the 8p23.1 duplication syndrome to date. ... The copy number of the adjacent repeats may also be altered. The 8p23.1 duplication syndrome cannot be distinguished using conventional cytogenetics from high level copy number variation of the repeats themselves. [1] [2] Both de novo cases and families with transmitted duplications from parents of both sex are known. ... (January 2008). "8p23.1 duplication syndrome; a novel genomic condition with unexpected complexity revealed by array CGH" .
8p23.1 duplication syndrome is a rare chromosomal anomaly syndrome, resulting from the partial duplication of the short arm of chromosome 8, with a highly variable phenotype, principally characterized by mild to moderate developmental delay, intellectual disability, mild facial dysmorphism (incl. prominent forehead, arched eyebrows, broad nasal bridge, upturned nares, cleft lip and/or palate) and congenital cardiac anomalies (e.g., atrioventricular septal defect).
Anonychia and its milder phenotypic variant, hyponychia, usually occur as a feature of genetic syndromes, in association with significant skeletal and limb anomalies. ... The findings added to evidence indicating that mesenchymal-epithelial interactions are crucial in nail development and put anonychia on the growing list of congenital malformation syndromes caused by Wnt signaling pathway defects.
Strandskov (1939) noted similarities between the nail phenotype of these patients and those of Osterreicher (1929); however, affected individuals in the latter pedigree who lacked thumbnails also tended to lack patellae (see nail-patella syndrome, 161200), whereas Strandskov (1939) stated that 'this does not appear to hold true for the affected members of the present pedigree.' Absent thumbnails in female members of 3 generations of a family proved on further study to represent the nail-patella syndrome (161200) (Schleutermann, 1968).
Isolated congenital anonychia is characterized by nail abnormalities ranging from onychodystrophy (dystrophic nails) to anonychia (absence of nails). Onychodystrophy-anonychia has been described in at least four generations of a family with male-to-male transmission, suggesting autosomal dominant transmission. Anonychia has been described in approximately less than 20 cases; it is likely to be transmitted as an autosomal recessive trait. Total anonychia congenita, in which all the fingernails and toenails are absent, may have an autosomal dominant inheritance pattern.
Description Although nails appear normal at birth, dystrophic changes develop within the first decade of life, resulting in onycholysis of fingernails and anonychia of toenails (summary by Rafiq et al., 2004). This disorder is referred to here as nonsyndromic congenital nail disorder-9 (NDNC9). For a list of other nonsyndromic congenital nail disorders and a discussion of genetic heterogeneity, see NDNC1 (161050). Clinical Features Rafiq et al. (2004) reported a 6-generation consanguineous Pakistani family with autosomal recessive transmission of a form of hereditary nail dysplasia. Affected individuals had normal nails at birth, but onychodystrophy began at age 7 or 8 and resulted in anonychia of the toenails (complete absence of nails) and onycholysis of the fingernails (wide separation of nail from nail bed and dystrophy of free margins).
PARC syndrome is a rare genetic developmental defect during embryogenesis syndrome characterized by the association of congenital poikiloderma (P), generalized alopecia (A), retrognathism (R) and cleft palate (C).
Verloes et al. (1990) described a possibly new autosomal dominant syndrome that combined poikiloderma, alopecia, retrognathism, and cleft palate--the PARC syndrome.
When hemangiomata are associated, the condition is known as Maffucci syndrome (614569). Clinical problems caused by enchondromas include skeletal deformity and the potential for malignant change to osteosarcoma (Schwartz et al., 1987). ... Population Genetics Sun et al. (1985) reported that 9 patients with Maffucci syndrome seen at the Mayo Clinic developed chondrosarcoma. ... In total, 35 of 43 (81%) individuals with Ollier disease and 10 of 13 (77%) with Maffucci syndrome carried IDH1 (98%) or IDH2 (2%) mutations in their tumors. ... Amary et al. (2011) analyzed 74 tumors from 40 individuals (32 with Ollier disease, 8 with Maffucci syndrome) for mutations in IDH1 (altering arg132) and IDH2 (altering arg140 and arg172). ... Amary et al. (2011) suggested a model in which IDH1 mutations are early post-zygotic events in individuals with these syndromes, implying that the mutations are required for tumorigenesis.
Enchondromatosis is a rare primary bone dysplasia disorder characterized by the development of multiple mainly unilateral or asymmetrically distributed enchondromas throughout the metaphyses of the long bones.
Ollier disease is a skeletal disorder characterized by multiple enchondromas , which are noncancerous (benign) growths of cartilage that develop within the bones. These growths may lead to skeletal deformities, limb discrepancy, and fractures. The enchondromas primarily occur in the limb bones, especially the bones of the hands and feet. They tend to develop near the ends of the bones, where growth occurs. Symptoms often appear in the first decade of life. The underlying cause of Ollier disease is not fully understood.
A related disorder called Maffucci syndrome also involves multiple enchondromas but is distinguished by the presence of red or purplish growths in the skin consisting of tangles of abnormal blood vessels (hemangiomas).
The clinical features somewhat resembled those of Smith-Lemli-Opitz syndrome (270400), which is caused by a defect in cholesterol biosynthesis. ... Herman (2003) reviewed the cholesterol biosynthetic pathway and 6 disorders involving enzyme defects in post-squalene cholesterol biosynthesis: Smith-Lemli-Opitz syndrome, desmosterolosis (602398), X-linked dominant chondrodysplasia punctata (CDPX2; 302960), CHILD syndrome (308050), lathosterolosis, and hydrops-ectopic calcification-moth-eaten skeletal dysplasia (HEM; 215140).
Differential diagnosis The main differential diagnosis is Smith-Lemli-Opitz syndrome (see this term) that shares many clinical features with lathosterolosis but that can be excluded with biochemical and genetic testing.
Acquired angioedema (AAE) is a rare disorder that causes recurrent episodes of swelling (edema) of the face or body, lasting several days. People with AAE may have swelling of the face, lips, tongue, limbs, or genitals. People with AAE can have edema of the lining of the digestive tract, which can cause abdominal pain and nausea, as well as edema of the upper airway, which can be life-threatening. Swelling episodes may have various triggers, such as mild trauma (such as dental work), viral illness, cold exposure, pregnancy, certain foods, or emotional stress. The frequency of episodes is unpredictable and can vary widely. There are two forms of AAE.
A number sign (#) is used with this entry because of evidence that susceptibility to angioedema induced by angiotensin-converting enzyme (ACE; 106280) inhibitors (AEACEI) is conferred by variation in the XPNPEP2 gene (300145) on chromosome Xq25. Description Approximately 40 million people take ACE inhibitors (ACEi) to treat hypertension and cardiovascular disease. A small proportion of white patients who take ACEi (0.1-0.7%) develop angioedema (AEACEI) (Israili and Hall, 1992; Vleeming et al., 1998), a potentially life-threatening side effect characterized by swelling of the face, lips, tongue, and airway that can lead to suffocation and death if severe. ACEi-associated angioedema is 4 to 5 times more prevalent among African Americans (Brown et al., 1996; Coats, 2002). Other risk factors include female sex, smoking, immunosuppressant therapy, and seasonal allergies.
The edemas may involve the digestive tract resulting in a clinical picture similar to that seen in intestinal occlusion syndrome, sometimes associated with ascites and hypovolemic shock. ... Type 1 AAE (see this term) is frequently associated with lymphoproliferative syndromes and accelerated consumption of C1-INH, and with autoimmune diseases that may manifest several years after the initial episodes of angioedema. ... Differential diagnosis The differential diagnosis should include intestinal occlusion syndrome, hereditary angioedema and histamine-induced angioedema (of allergenic or nonallergenic origin) generally associated with urticaria.
Microcephaly-cardiomyopathy syndrome is characterised by severe intellectual deficit, microcephaly and dilated cardiomyopathy. Hand and foot anomalies have also been reported. The syndrome has been described in three individuals.
In a brother and sister of Afrikaner stock, Winship et al. (1991) observed severe microcephaly with mental retardation and dilated cardiomyopathy developing in infancy and later resolving completely. Both had bilateral fifth finger clinodactyly and sandal gaps on both feet. Kennedy et al. (1999) reported a 9-year-old Canadian girl with features similar to those of the sibs in the report of Winship et al. (1991). She was born to nonconsanguineous parents following an uncomplicated pregnancy. The neonatal period was complicated by seizures and cardiac failure secondary to a ventricular septal defect.