-
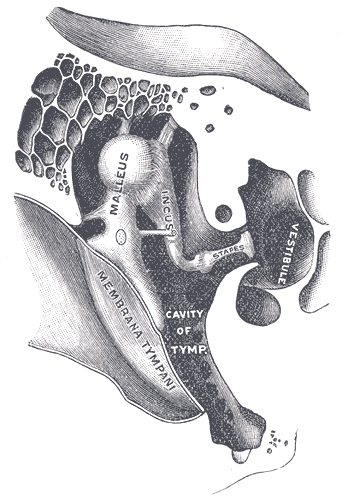
Otosclerosis
Wikipedia
Howard Hughes the pioneering American aviator, engineer, industrialist, and film producer also suffered from otosclerosis. [27] Frankie Valli , lead singer of The Four Seasons , suffered from it in the late 1960s and early 1970s, forcing him to "sing from memory" in the latter part of the 70s (surgery restored most of his hearing by 1980). [28] Pittsburgh Penguins forward Steve Downie suffers from otosclerosis. [29] The British queen Alexandra of Denmark suffered from it, leading to her social isolation; Queen Alexandra's biographer, Georgina Battiscombe , was able to have "some understanding of Alexandra's predicament" because she too had otosclerosis. [30] [31] Adam Savage , co-host of MythBusters , uses a hearing aid due to otosclerosis. [32] Sir John Cornforth , Australian-British Nobel Prize in Chemistry laureate [33] References [ edit ] ^ " otosclerosis " at Dorland's Medical Dictionary ^ Uppal, S.; Bajaj, Y.; Rustom, I.; Coatesworth, A. ... PMID 19205157 . ^ "Use of bisphosphonates for otosclerosis" Archived 2014-10-28 at the Wayback Machine , Fresh Patents. ^ Chris De Souza, Michael E.COL1A1, RELN, COL1A2, GDF6, BPTF, TGFB1, PSMD12, ZNF383, OTSC2, TNFRSF11B, NOG, OTSC1, TNF, CD46, HLA-A, BMP4, BMP2, BTF3P11, SERPINF1, REN, STRC, ZNF410, MEPE, OTSC4, VDR, OTSC3, OTSC7, THEMIS, SP1, OTSC5, AGT, MAPK3, PON1, ANGPT2, VPS51, CP, ACE, SLC26A2, GC, GH2, HLA-B, HLA-C, HMGB1, HNF4A, IGF1, IL6, ITGAV, AGTR1, OTSC10
-
Corneal Opacity
Wikipedia
Depending on type and density of corneal opacity different types of keratoplasty may be used such as: [28] Penetrating keratoplasty: It is the traditional full thickness corneal transplant procedure, in which trephine (a circular cutting device) is used to cut opaque cornea, a similar-sized portion of the donor cornea is removed with a second trephine. ... "New Surgical Strategy for Corneal Tattooing Using a Femtosecond Laser". Cornea . 28 (1): 80–4. doi : 10.1097/ICO.0b013e318181a83c .GLB1, WDR37, SLC4A4, JAG1, CRYAA, B3GALNT2, BAZ1B, POMT1, YAP1, RXYLT1, CEP57, GTF2IRD1, PIGL, PEX16, TRIP13, LARGE1, XPR1, BUB3, PEX11B, POLR3A, SUMF1, ELP1, IKBKG, PEX3, CLIP2, TBCE, TAT, SLC20A2, RFC2, REV3L, B4GAT1, ATOH7, B3GLCT, MCOLN1, GJB4, TWIST2, POMGNT2, POMK, SLC4A11, FRAS1, SRD5A3, FKRP, PORCN, MYORG, NGLY1, PLXND1, PEX26, POMGNT1, FERMT1, ATAD3A, RIPK4, MBTPS2, POMT2, TBL2, PIGN, ALDH18A1, ABCA1, PEX5, PEX2, CRYGD, GJB3, GJA8, GJA1, KDSR, FUCA1, FLNB, FGFR1, FKTN, ELN, DAG1, CYP1B1, CRYGC, PEX19, CRYBB2, CRYBB1, CRYBA4, COL11A1, COL8A2, COL4A1, BUB1B, BUB1, ASAH1, STS, APOA1, GLA, GTF2I, GUSB, IDUA, CTSA, PIK3R1, PEX14, PEX13, PEX12, PEX10, PEX6, PEX1, PDGFRB, PDGFB, PAX6, OCRL, NF2, NEU1, MMP14, MAF, LTBP2, LRP5, LIMK1, LCAT, KRAS, KIF11, CRPPA, TGFBI, RSPO1

-
Athetosis
Wikipedia
Clin. Obstet. Gynecol . 51 (4): 816–28. doi : 10.1097/GRF.0b013e3181870ba7 . ... Clinical Obstetrics and Gynecology . 51 (4): 816–28. doi : 10.1097/GRF.0b013e3181870ba7 .ACO2, NDE1, WARS2, CUX2, PIGN, PHGDH, SLC17A5, UQCRQ, UBQLN2, APTX, SMPD1, FOXRED1, PIGV, NGLY1, TWNK, TRAPPC11, UBA5, PRRT2, SUCLA2, SLC20A2, KIF1A, GAMT, AUH, CACNA1A, CACNA1D, CYB5A, CYB5R3, EPRS1, FOXG1, GNAO1, SLC16A2, KCNA1, KIF5A, PDGFB, PDGFRB, ALDH18A1, SLC6A8, SLC46A1, NPHS1

-
Arteriovenous Malformation
Wikipedia
Miller was diagnosed with AVM after filming Yogi Bear in New Zealand in 2010; Miller described his experience with the disease on the Pete Holmes podcast You Made It Weird on October 28, 2011, shedding his comedian side for a moment and becoming more philosophical, narrating his behaviors and inability to sleep during that time. ... He still records and performs to this day. [28] YouTube vlogger Nikki Lilly (Nikki Christou), winner of the 2016 season of Junior Bake Off was born with AVM, which has resulted in some facial disfigurement. [29] Composer and lyricist William Finn was diagnosed with AVM and underwent Gamma Knife surgery in September 1992, soon after he won the 1992 Tony Award for best musical, awarded to " Falsettos ". [30] Finn wrote the 1998 Off-Broadway musical A New Brain about the experience.RASA1, AKT1, PTEN, PIK3CA, TEK, NOTCH1, FANCF, RFWD3, FANCI, ARHGAP31, FANCG, FANCE, FANCB, GUSB, FANCD2, FANCC, FANCA, DOCK6, FANCL, RBPJ, DLL4, MNX1, ERCC4, UBE2T, MAD2L2, ADAMTS3, XRCC2, VHL, VEGFA, RAD51, RAD51C, SLPI, FANCM, GNAQ, BRCA1, EOGT, ENG, CCBE1, FAT4, ACVRL1, PALB2, ARL6IP6, CBS, BRCA2, BRIP1, SLX4, SMAD4, IL6, TNF, KRAS, APOE, EPHB4, SMS, ANGPTL4, IL1B, GDF2, RSS, BRAF, ANGPT2, MAP2K1, MMP9, SOX17, PIK3CB, PIK3CD, PIK3CG, VWF, ANXA5, AGT, VCAM1, MMP3, MMP2, PART1, ICAM1, ELN, F3, EDN1, ACE, CLN3, CD6, SMG8, KDR, ABCA2, PAGE4, SYNGAP1, TGFB2, TGFB3, TGFBR1, THBS1, MIR137, SMUG1, TLR4, PDGFD, NES, COL18A1, CDKN2B-AS1, MIR210, PIK3R4, MIR195, KLF2, TMEM100, ESAM, BMP10, TGFB1, MVP, NOS1AP, MIR18A, PAGE1, BLNK, NOTCH3, STAT3, DLAT, MTOR, FN1, FLT1, FOXC2, FGF2, F2, ETS1, EPHB2, EFNB2, EFNA1, EDNRA, CRYAB, SPP1, COX8A, CDKN2A, CDH5, CD34, CCT, CASP3, VPS51, BDNF, ANPEP, ALX3, ALCAM, GJA5, HIF1A, HRAS, IFNG, SOX2, SNCA, SLC2A1, S100B, RBBP8, PTPN14, MAPK1, PLOD2, PECAM1, PDGFRB, PCYT1A, NOTCH4, ACTB, NOS3, NOS2, NOS1, MYB, MSH3, ABCC1, MGP, MDM4, MDM2, ITGB8, CERNA3

-
Pheochromocytoma
Omim
Mutation in the KIF1B Gene In a pheochromocytoma tumor sample and in germline DNA from the corresponding patient, Schlisio et al. (2008) identified a mutation in the KIF1B gene (S1481N; 605995.0005). The proband was a 28-year-old female who presented at 17 months of age with a neuroblastoma and in adulthood developed a mature ganglioneuroma and bilateral pheochromocytoma. ... These findings suggested that NF1 loss of function is a frequent event in the tumorigenesis of sporadic pheochromocytoma. Modifier Genes In 1 of 28 sporadic pheochromocytomas, Woodward et al. (1997) identified a mutation in the glial cell line-derived neurotrophic factor gene (GDNF; R93W; 600837.0001), which is a natural ligand for RET.RET, MAX, VHL, TMEM127, SDHB, SDHD, GDNF, KIF1B, TP53, SDHC, CHGA, TH, NF1, DBH, EGLN1, DDC, COMT, MAOA, MAOB, DMBT1, CCND1, FH, DLST, NGF, POMC, SST, PRKAR1A, SARDH, SORD, CALCA, SDHA, EPAS1, SDS, ADM, SDHAF2, CRYGD, VEGFA, PNMT, CDKN1B, MEN1, SMUG1, HIF1A, MYC, NPY, IGF2, MDH2, STMN1, IL6, PMPCA, HSP90AA1, NTRK1, SSTR2, CRH, TERT, BCL2, CDKN2A, SCARB1, HRAS, SLC6A2, APP, UTS2, MTOR, REN, FLT1, KIT, SNCA, ADCYAP1, HTC2, SCG2, EPO, MT1A, PIK3CG, PIK3CD, PIK3CB, PIK3CA, DRD2, PENK, EGLN3, RASSF1, SYP, IDH2, HSPA4, IGF1, IGF1R, TERC, PHOX2B, TNF, DCTN6, UCP1, GALR2, PTPRF, PSMD9, VIPR1, PRDM2, BAP1, MKI67, ZNRD2, H3P10, EGLN2, BRAF, MIB1, EGF, FGF21, CPE, CHGB, TICAM2, CASP3, MIR183, FGFR1, MIR210, AGTR2, AGT, PARP1, TMED7-TICAM2, RAB4B-EGLN2, H3P23, EPOR, IFI27, SETD2, GSK3B, TMED7, GHRL, GAL, ADIPOR1, UTS2R, GIPC1, VIP, TTR, TWIST1, TXN, VBP1, MIR15A, MALAT1, MIR22, ANGPTL4, VIPR2, ZACN, SOX2-OT, HSD17B13, H19, HYLS1, KMT2D, GPR68, STAR, MIR96, TRPC6, PDE11A, PGR-AS1, SYT1, CD274, ELOB, ELOA, HNF1B, QPCT, MIR1225, TGFA, CNTN4, MIR541, CDKN2B-AS1, MIR765, TGM2, POTEF, TIMP2, TIMP3, MIR374A, FSIP1, TDRD9, MEG3, SDHAF3, BMS1, DGCR2, HDAC6, ABCB6, DNM1L, PTPRU, SLC17A6, ACKR3, KRT20, RETN, MZB1, FARP1, RNF41, CALCRL, CTNNBL1, MPZL2, RAMP2, CIB1, HPSE, MAML3, RGS6, DHRS11, GDE1, ADIPOQ, UCN3, CUL4A, CUL2, FNDC3A, CBX4, PDE8B, NCOA1, PDE5A, HSD17B7, CGB8, ARTN, AIP, CGB5, CLDN10, RASSF5, DIRAS3, TET1, ARHGEF1, ADIPOR2, RWDD3, ACACA, SSTR5, EDNRA, EGFR, EMP3, ENPEP, EPHA3, ERBB2, ERV3-1, ESRRA, ETS1, FGFR3, FOXM1, FN1, G6PD, GAP43, GATA3, GATA4, GCG, GCGR, GHRHR, GLP1R, GNAS, GNRH1, NR3C1, GUSB, HCRTR1, HIC1, HPR, HTR4, IDH1, IFNG, EDNRB, EDN1, SSTR1, ACE, ACTB, ACVR1, ACVR1B, ADCYAP1R1, AGRP, AGTR1, AKT1, APC, BIRC5, APRT, ASCL1, ATRX, BDNF, BMP7, CA9, CASP8, CAT, SERPINH1, CBS, MS4A1, CDC42, CDH1, CDKN2C, CGA, CGB3, COL11A2, CRP, CS, MAPK14, IGFBP1, IGFBP2, IL1A, IL5, PKD2, PLAGL1, PMCH, ACO1, PPARG, MAPK3, MAPK8, PTEN, PTH, PTGS2, RAB6A, RAC1, RASA1, RBP3, REG1A, RPS6KB1, RPS6KB2, S100A12, SCTR, CCL7, CX3CL1, CXCL12, SIM2, SLC6A3, SLC18A2, SMPD1, SNAI1, SNAP25, SRC, SLC25A3, ABCB1, PDYN, MITF, INSR, IRF3, ISL1, KCNJ5, KDR, KRT6B, KRT17, L1CAM, LEP, LEPR, LGALS3, MFAP1, MGMT, MLH1, SLC26A4, MMP9, MOS, MSH2, MYCL, NFE2L2, NOTCH1, NRTN, NTS, GPR143, PCDHGC3, PCSK2, PDC, PDGFRB, POU4F1

-
Congenital Mirror Movement Disorder
Wikipedia
Therefore, quality of life can be severely hampered. [3] CMM disorder’s prevalence in the world is thought to be less than 1 in 1 million people. [1] [5] Because of its rarity, researchers suggest that some mildly affected individuals may never be diagnosed. [2] [6] It is important not to confuse congenital mirror movement disorders, a rare genetically based neurologic disease, with acquired mirror movement disorders that present themselves during one’s lifetime due to other reasons (stroke for example). [2] Contents 1 Causes 2 Pathophysiology 2.1 Interhemispheric connections 2.2 Motor cortex 2.3 Corticospinal tract 3 Diagnosis 4 Treatment and Management 5 Related Diseases 6 References 7 External links Causes [ edit ] The specific molecular mechanism that underpins this movement disorder is not well known. [2] However, most researchers suggest that it follows an autosomal dominant genetic inheritance pattern in which mutations in certain genes give rise to structural abnormalities in nervous system networks responsible for voluntary skeletal muscle movement , which, in turn, result in the functional movement abnormalities seen in patients. [1] [2] [7] [8] [9] Despite being autosomal dominant, it is important to note that the disease has variable expressivity . [3] That is, patients who have inherited a mutated dominant allele, along with their genetically affected parent, can be symptomatic or asymptomatic for CMM disorder. [4] The genes that currently have evidence to be associated with CMM disorder include DCC (deleted in colorectal carcinoma), DNAL4 (dynein axonemal light chain 4), and RAD51 (recombination protein A) . [6] [10] DCC encodes a receptor for NTN1 (netrin-1), a protein thought to be responsible for axon guidance and neuronal cell migration during development . [11] [12] A mutation of this gene (including nonsense , splice site mutation , insertions , frameshift ) has been identified as a possible cause for CMM disorder. [2] [13] [14] Experiments in mice also support the claim that CMM disorder is associated with genetic mutations in DCC . [9] Kanga mice, lacking the P3 intracellular domain of the DCC receptor, show a hopping gait, moving their hind legs in a strictly paired fashion, as do kangaroos . [3] [15] DNAL4 encodes a component of dynein motor complex in commissural neurons of the corpus callosum . [1] [6] [3] In contrast to DCC , DNAL4 is thought to have a recessive inheritance pattern for the CMM disorder. [8] In CMM disorder patients, researchers found splice site mutations on DNAL4 , which caused skipping of exon 3, and thereby omission of 28 amino acids from DNAL4 protein. [8] This mutant DNAL4 protein, in turn, could lead to faulty cross-hemisphere wiring, resulting in CMM. [8] [16] RAD51 maintains genome integrity by repairing DNA double-strand breaks through homologous recombination . [7] RAD51 heterozygous mutations, specifically premature termination codons , have been found in many CMM disorder patients through genome-wide linkage analysis and exome sequencing . [1] [2] [4] [6] In a mouse model, researchers also found RAD51 products in corticospinal tract axons at the pyramidal decussation . [7] They therefore suggest that RAD51 might be a gene that, when haploinsufficient, causes CMM disorder in humans. [7] Despite identification of three prospective genes, no genotype - phenotype correlations have yet been found. [1] [2] That is, the severity of clinical signs and symptoms does not correlate with the type of genetic variant. [3] [17] Mutations in the above genes account for a total of about 35 percent of cases. [1] Mutations in other genes that have not been identified likely account for the other cases of this disorder. [1] Pathophysiology [ edit ] There are three main pathophysiological hypotheses for congenital mirror movement disorder that exist. ... This might provide an alternate explanation for the presence of mild mirror movements in normally developing young children that typically disappear before the age of 7. [24] Some researchers propose that DCC mutations cause a reduction in gene expression and less robust midline guidance , which may lead to a partial failure of axonal fiber crossing and encourage development of an abnormal ipsilateral connection. [7] This is confirmed by other researchers who demonstrate that patients with DDC mutants show an increased proportion of ipsilateral axonal projections, and show that even a very small number of aberrant ipsilateral descending axons is sufficient to induce incorrect movement patterns. [11] [14] [25] These findings are corroborated by evidence from mice models, Kanga mice with a deletion of DCC , whose CST has been shown not to be altered , but rather partially rerouted ipsilaterally. [16] Diagnosis [ edit ] Currently, clinical diagnosis of CMM disorder has been based on clinical findings or molecular genetic testing . [2] Clinical Findings (Signs and Symptoms) [1] [2] [10] [26] [14] : onset of mirror movements in infancy or early childhood persistence of mirror movements into and throughout adulthood with the absence of other neurologic disorders little improvement nor deterioration of mirror movements over the course of one’s life intensity of mirrored movements increasing with the complexity of the voluntary movement involuntary mirror movements that are generally of lesser amplitude compared with voluntary movements predominant mirror movement in upper limbs, with increasing severity in more distal appendages (fingers) inability to perform tasks requiring skilled bimanual coordination occasional pain in the upper limbs during prolonged manual activities occasional observed subclinical mirroring movement, but detectable with accelerometer gloves Molecular genetic testing [1] : identification of a heterozygous mutant DCC, DNAL4, or RAD51 gene ( single gene test or multi-gene panel) Treatment and Management [ edit ] CMM has clear severe impacts on a patient’s ability to carry out daily manual tasks . [27] [17] It is recommended that children be placed under more forgiving school environments, allowing more time for written evaluations and limiting handwritten assignments, to ease the burden of the movement disability. [1] [3] Furthermore, because of patients’ inability to perform pure unilateral movements and their difficulty with tasks requiring skilled bimanual coordination, young and new members to the workforce are encouraged to consider professions that do not require complex bimanual movements, repetitive or sustained hand movements, or extensive handwriting, to reduce overuse, pain, and discomfort in upper limbs. [2] [5] Because of its pronounced and obviously noticeable signs and symptoms, CMM patients can suffer social stigma ; however, physicians need to make it clear to parents, family, and friends that the disorder bears no relation to intellectual abilities . [5] [28] However, the rarity of this neurologic disease, found in one in a million people, makes its societal and cultural significance quite limited. [6] Related Diseases [ edit ] Movement disorders Chiari malformation Klippel-Feil Syndrome Dystonia Cerebral palsy Parkinson's disease Epilepsies Amyotrophic lateral sclerosis Kallman's syndrome Alien hand syndrome Obsessive compulsive disorder Schizophrenia Congenital hemiplegia Moebius syndrome Seckel syndrome Wildervanck syndrome Polymicrogyria References [ edit ] ^ a b c d e f g h i j k l m n o Reference, Genetics Home.CDH2, DCC, RAD51, ANOS1, DNAL4, NTN1, FGFR1, FGF8, FEZF1, CCDC141, PROKR2, KISS1R, SPRY4, PROK2, KNL1, WDR11, CHD7, IL17RD, NSMF, FLRT3, CEP152, LINC01917, SEMA3A, CREBBP, HS6ST1, FGF17, HESX1, TACR3, SOX10, DUSP6, EP300, RAD51B, GDF6, MEF2C, SMN2, SCN8A, POMK, SMN1, NTNG1

-
Zttk Syndrome
Wikipedia
. ^ "Periventricular heterotopia | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov . Retrieved 2019-04-28 . ^ a b "Arnold Chiari Malformation: Symptoms, Types, and Treatment" . WebMD . Retrieved 2019-04-28 . ^ a b Vissers, Lisenka E. L. M.; Gilissen, Christian; Veltman, Joris A. (2015-10-27).
-
Aplastic Anemia
Wikipedia
This is referred to as idiopathic aplastic anemia and accounts for 75% of cases. [22] This compromises the effectiveness of treatment since treatment of the disease is often aimed at the underlying cause. [24] Those with a higher risk for aplastic anemia include individuals that are exposed to high-dose radiation, exposed to toxic chemicals, take certain prescription drugs, have pre-existing autoimmune disorders or blood disease, or are pregnant. [25] The five-year survival rate is higher than 75% among recipients of blood marrow transplantation. [24] Other treatment strategies include medications and blood transfusions. [25] Patients that are untreated will often die within a year as a result of the disease due to related complications, which are most commonly bleeding and infection due to deficiency of platelets and white blood cells, respectively. [24] There is not a screening test that currently exists for early detection and diagnosis of aplastic anemia. [22] Notable cases [ edit ] Marie Curie [26] Eleanor Roosevelt [27] Donny Schmit [28] Ted DeVita [29] Demetrio Stratos [30] John Dill (British Field Marshal) [31] See also [ edit ] Fanconi anemia Acquired pure red cell aplasia References [ edit ] ^ a b Young, Neal S. (2018-10-25). ... Retrieved December 10, 2012. ^ "AMA Motorcycle Museum Hall of Fame | Donny Schmit" . www.motorcyclemuseum.org . ^ "Oncologist Discusses Advancements In Treatment And The Ongoing War On Cancer" . NPR.org . October 28, 2015. ^ Pavese, Antonella (22 October 2006).IFNG, TERT, NBN, SBDS, PRF1, CSF3, CSF2, KIT, PIGA, RASA3, TERC, SRP72, TINF2, HLA-DRB1, TNF, DNAJC21, EFL1, RPL5, HOXA11, NHP2, ACD, NOP10, ELANE, DIPK1A, TCIRG1, SRP54, GFI1, PALB2, THPO, GATA2, CD34, RBM45, TP53, FOXP3, CD59, IL6, CDR3, STAT3, RPS19, IL10, GSTT1, HLA-A, CD55, GSTM1, TBX21, CD247, TGFB1, HLA-B, PDCD1, CXCL8, GEM, SH2D1A, GATA3, ID4, MPL, HPGDS, GPI, HLA-DQA1, MSN, KIR3DL1, DKC1, NRAS, FCGR3B, FAS, FASLG, ITGA2B, MIR204, IL27, STAT4, CXCL10, ASXL1, CXCR4, IL17A, CD48, HAVCR2, TNFSF10, CEACAM8, CSF3R, PPARG, CTLA4, MYDGF, NLRP2, NQO1, VEGFA, IL17D, IL23A, FCGR3A, YWHAE, WT1, ALDH2, VDR, UROD, TRAF1, THBD, TGFBI, TFR2, TERF2, TERF1, TRB, TAL1, SPARC, SNCA, TFRC, BRD4, LOH19CR1, DGKZ, QRSL1, HAMP, IL21, BCORL1, NHEJ1, SLCO6A1, PPARGC1B, IL23R, HBFQTL4, APLF, FOLH1B, GSTK1, MIR126, MIR34A, CCR2, RPL17-C18orf32, KLRC4-KLRK1, TET2, SCLY, WT1-AS, CAP1, CD164, SPHK1, HACD1, MSC, SPATA2, GDF11, ECI2, SORBS1, SLC25A37, KLRK1, SF3B1, SELPLG, SH2B1, PTPN22, CD274, ANAPC2, SMARCA1, NFE2L2, SELP, F9, CSF1R, CTAA1, CYP1A1, CYP2B6, CYP2D6, CYP2E1, EGF, ETV6, EZH2, FANCA, GSTP1, FANCD2, FANCG, FGF1, FGF2, FLT1, FOLH1, GABPA, GATA1, CBLIF, CCR6, TPP1, CDKN2D, CDKN2A, AR, SERPINC1, B2M, BCL2, BGLAP, BMP4, BMP6, BRCA2, ZFP36L1, BTK, CA1, RUNX1, CBLB, CD3G, CD19, CD27, TNFRSF8, CD40LG, CD70, CXCR3, GYPA, CCL20, PMS1, SMAD4, MDM4, NCAM1, NFATC2, APC, NOS2, PAFAH1B1, SLC26A4, SERPINB6, PRTN3, GYPB, PTGDS, RAG2, REG1A, RMRP, RPL17, S100A8, S100A9, SCT, CCL2, LNPEP, LGALS9, LEPR, LEP, GYPE, HFE, HLA-DQB1, HOXB4, HP, HES1, ICAM1, IGH, IL1B, IL2, IL3, IL4, IL11, IL15, IL18, ITGB3, KIR3DL2, KRT7, KTN1, H3P13
-
Folate Deficiency
Wikipedia
Prevention and treatment [ edit ] Diet. [ edit ] Folate is acquired in the diet by the consumption of leafy green vegetables, legumes and organ meats. [27] When cooking, use of steaming , a food steamer , or a microwave oven can help keep more folate content in the cooked foods, thus helping to prevent folate deficiency. [28] [29] [30] Supplementation [ edit ] Folic acid is a synthetic derivative of folate and is acquired by dietary supplementation. [21] Multi-vitamin dietary supplements contain folic acid as well as other B vitamins . ... PMID 27664775 . ^ a b c d Lassi, Zohra S; Salam, Rehana A; Haider, Batool A; Bhutta, Zulfiqar A (28 March 2013). "Folic acid supplementation during pregnancy for maternal health and pregnancy outcomes".DHFR, MTHFR, CBS, MTR, BCL2, PCBP1, SLC46A1, MTRR, IL2, PAX3, TAGLN, NOTCH1, PCNA, MYOG, PSEN1, QDPR, RFC1, ROS1, SHMT1, MYC, SLC19A1, ADH5, TBX1, TCN2, TYMS, H3-4, SQSTM1, KHDRBS1, DKK1, FAM215A, NUP62, DCTN4, WLS, SERPINA13P, MIRLET7G, MIR34A, CBSL, TGFB1, MTHFD1, ALDH2, FHIT, APOE, BRCA1, BRCA2, C5, C5AR1, CRP, CSTB, CTSL, CYP2J2, DNMT1, DVL1, MARK2, ESR1, FOLR1, LPL, FPGS, GLI2, GNAS, GPT, GTF2H1, GZMM, H2AX, HSPA5, IFNG, IGF1, IGF2, IL10, LEP, RN7SL263P

-
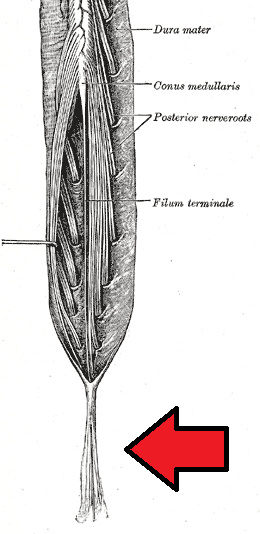
Cauda Equina Syndrome
Wikipedia
CES is often concurrent with congenital or degenerative diseases and represents a high cost of care to those admitted to the hospital for surgery. [11] [25] Hospital stays generally last 4 to 5 days, and cost an average of $100,000 to $150,000. [25] Delays in care for cauda equina results in the English NHS paying about £23 million a year in compensation. [26] Other animals [ edit ] Degenerative lumbosacral stenosis (DLSS), also known as cauda equina syndrome, is a pathologic degeneration in the lumbosacral disk in dogs; affecting the articulation, nerve progression, tissue and joint connections of the disk. [27] [28] This degeneration causes compressions in soft tissues and nerve root locations in the ultimate caudal area of the medulla , causing neuropathic pain in the lumbar vertebrae . [29] [30] References [ edit ] ^ a b c d e f g h i j k l m n Gardner A, Gardner E, Morley T (May 2011). ... "Surgical treatment of degenerative lumbosacral stenosis in dogs". Veterinary Surgery . 28 (2): 91–8. doi : 10.1053/jvet.1999.0091 .
-
Charcot-Marie-Tooth Disease, X-Linked Dominant, 1
Omim
Genotype/Phenotype Correlations Shy et al. (2007) evaluated 73 male patients with CMTX1, ranging from 9 to 76 years of age, who had a total of 28 distinct GJB1 mutations. Two patients had a complete deletion of the GJB1 gene, and all others had truncating or missense mutations affecting various regions of the protein. ... Population Genetics Abe et al. (2011) identified GJB1 mutations in 19 (8.5%) of 227 Japanese patients with demyelinating CMT and in 6 (4.7%) of 127 Japanese patients with axonal CMT. In 28 (5.3%) of 527 unrelated Korean families with CMT, Kim et al. (2012) identified 23 different mutations in the GJB1 gene (see, e.g., 304040.0005 and 304040.0011).
-
Acute Generalized Exanthematous Pustulosis
Wikipedia
"Acute generalized exanthematous pustulosis: clinical characteristics, etiologic associations, treatments, and outcomes in a series of 28 patients at Mayo Clinic, 1996-2013". ... Retrieved 25 October 2020 . ^ Ropars, Nolwenn; Darrieux, Laure; Tisseau, Laurent; Safa, Gilles (2014-09-28). "Acute generalized exanthematous pustulosis associated with primary Epstein-Barr virus infection" .

-
Lewy Body Dementias
Wikipedia
McKeith , professor and researcher of Lewy body dementias, commented that Williams' symptoms and autopsy findings were explained by dementia with Lewy bodies. [22] The British author and poet Mervyn Peake died in 1968 and was diagnosed posthumously as a probable case of DLB in a 2003 paper published in JAMA Neurology . [23] Sahlas said his death was "variously ascribed to Alzheimer disease, Parkinson disease, or postencephalitic parkinsonism". [23] Based on signs in his work and letters of progressive deterioration, fluctuating cognitive decline, deterioration in visuospatial function, declining attention span , and visual hallucinations and delusions, his may be the earliest known case where DLB was found to have been the likely cause of death. [23] Other entertainers and artists who have or died from LBD include Estelle Getty , an actress known for her role in the television series The Golden Girls , [24] Nicholas King , a US actor and horticulturist, [25] actress Dina Merrill , [26] Donald Featherstone , who created the plastic pink flamingo, [27] American radio and television host Casey Kasem , [28] Canadian singer Pierre Lalonde , [29] [30] and graphic artist/film set designer Ron Cobb . [31] Individuals from industry or government who have or died from LBD are Seymour Berry , US Director of the Bureau of Engraving and Printing , [32] Los Angeles Times publisher Otis Chandler , [33] Philip J. ... Retrieved April 19, 2018 . ^ Deerwester J (September 28, 2018). "Ted Turner has Lewy Body Dementia" .
-
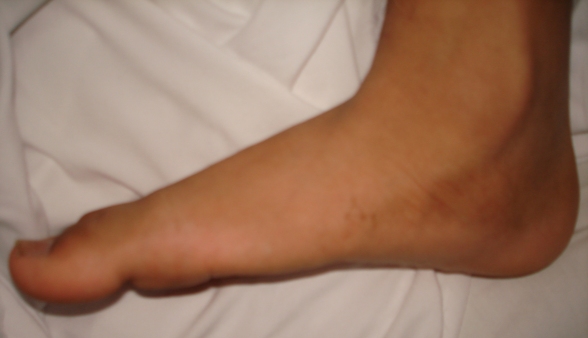
Flat Feet
Wikipedia
University of South Australia, Spencer Gulf Rural Health School. 28 (6): 715–23. doi : 10.3113/FAI.2007.0715 . ... Part 1: Prospective cohort study" . Military Medicine . 170 (6): 623–28. doi : 10.7205/MILMED.170.7.623 .HOXD10, ELN, AUTS2, B4GALT7, EXOC6B, PHF8, IQSEC2, DHX30, CNKSR2, PUF60, AP4S1, ADNP, IL1RAPL1, PRDM5, MID2, TLK2, RAI1, STAG2, MED13L, AP4E1, RIN2, ATP6V0A2, FLRT3, FTSJ1, RAB3GAP2, NSMF, B3GAT3, BSCL2, TBL2, UBE2T, ANKRD11, GMPPB, ZDHHC9, RLIM, DACT1, AP4B1, ABL1, DEAF1, CCNK, AP4M1, AIFM1, BAZ1B, HERC1, FGF17, HESX1, CDK10, TRIP4, OFD1, CUL4B, USP9X, GDF5, GAN, MFAP5, CRLF1, EFTUD2, SEC23A, AMMECR1, MAD2L2, SEMA3A, ATP6AP2, HUWE1, SCO2, MED12, CUL7, LONP1, FRMPD4, GTF2IRD1, TBX4, CHST3, ARHGEF6, HS6ST1, DPM3, IL17RD, ALMS1, SLC9A7, C12orf57, STX1B, GORAB, RFT1, SLC39A13, ALG2, MYPN, CANT1, KISS1R, ZNF469, SLX4, ADGRV1, BRIP1, SPRTN, RAB39B, B3GALT6, PHIP, CCDC141, GDF6, USP27X, FEZF1, PTPRQ, HCN1, ZNF81, BRWD3, PROKR2, ALG14, ARX, VPS13B, PTCHD1, NDUFAF6, TANGO2, RAB33B, LAS1L, SPRY4, IARS2, FANCM, CHD8, SELENON, SALL4, HDAC8, WDR11, PACS1, TRIM8, CHD7, SLC29A3, FANCI, RFWD3, FANCL, FKBP14, WDR19, TRPV4, PROK2, PIEZO2, CXorf56, XYLT1, XYLT2, NSD1, SIL1, REEP1, UPF3B, PALB2, EHMT1, ALG13, PYROXD1, FBXO11, ASXL3, RAB7A, ZNF148, ACTA2, BPTF, FGF8, FGD1, FBN1, FANCG, FANCF, FANCB, ACSL4, FOXG1, FANCE, FANCD2, FANCC, FANCA, EXT2, ERCC4, FGFR1, FOXE3, GRM1, GABRG2, GRIA3, GLI3, GFPT1, GDI1, GATA4, GARS1, GABRD, FLI1, MTOR, FN1, FMR1, FLNB, FLNA, FLII, EP300, DUSP6, DPAGT1, ATR, CAPN1, CACNA1A, BRCA2, BRCA1, BCR, ATRX, ATP7A, DMD, ATP6V1E1, ATP6V1B2, AHCY, AGTR2, AGA, AEBP1, CTSC, CHAT, CLCN4, COL1A1, COL1A2, COL2A1, COL5A1, COL5A2, COL6A1, COL6A2, COL6A3, COL12A1, CPT2, CREBBP, CRKL, DCC, DLG3, GRIN2D, GTF2I, ZNF41, SCN2A, TACR3, SYP, SOX10, SLC16A2, SKI, SCN9A, SCN1B, TGFB1, SCN1A, SBF1, SARS1, SALL1, RYR1, RPS6KA3, TCF4, TGFB2, HCFC1, TTN, ZNF711, XRCC2, CLIP2, VLDLR, UCHL1, UBE2A, TRPS1, TGFB3, TRIO, TPM3, TPM2, TSPAN7, TGFBR2, TGFBR1, RFC2, CHCHD10, RAD51C, LMNA, MGAT2, MECP2, MAT2A, SMAD3, LOX, LMX1B, LIMK1, RAD51, LIFR, KIF22, KCNH1, ANOS1, HSPG2, HNRNPH2, MMP2, MYH7, MYH11, MYLK, NONO, PAK3, CHMP1A, PEX1, PEX6, PLOD1, PMM2, PRKG1, MAPK1, PTCH1, PURA, PYCR1, ALDH18A1, PDPN
-
Childhood Cancer
Wikipedia
Premature heart disease is a major long-term complication in adult survivors of childhood cancer. [23] Adult survivors are eight times more likely to die of heart disease than other people, and more than half of children treated for cancer develop some type of cardiac abnormality, although this may be asymptomatic or too mild to qualify for a clinical diagnosis of heart disease. [23] Childhood cancer survivors are also at risk of developing adverse effects on the kidneys [24] and the liver. [25] The risk of liver late adverse effects in childhood cancer survivors is increased in those who have had radiotherapy to the liver and in people with factors such as higher body mass index and chronic viral hepatitis. [25] Certain treatments and liver surgery may also increase the risk of adverse liver effects in childhood cancer survivors. [25] Epidemiology [ edit ] Internationally, the greatest variation in childhood cancer incidence occurs when comparing high-income countries to low-income ones. [26] This may result from differences in being able to diagnose cancer, differences in risk among different ethnic or racial population subgroups, as well as differences in risk factors. [26] An example of differing risk factors is in cases of pediatric Burkitt lymphoma , a form of non-Hodgkin lymphoma that sickens 6 to 7 children out of every 100,000 annually in parts of sub-Saharan Africa, where it is associated with a history of infection by both Epstein-Barr virus and malaria . [26] [27] [28] In industrialized countries, Burkitt lymphoma is not associated with these infectious conditions. [26] US [ edit ] In the United States, cancer is the second most common cause of death among children between the ages of 1 and 14 years, exceeded only by accidents. [20] More than 16 out of every 100,000 children and teens in the U.S. were diagnosed with cancer, and nearly 3 of every 100,000 died from the disease. [20] In the United States in 2012, it was estimated that there was an incidence of 12,000 new cases, and 1,300 deaths, from cancer among children 0 to 14 years of age. [29] Statistics from the 2014 American Cancer Society report: Ages birth to 14 [30] Sex Incidence Mortality Observed Survival % Boys 178.0 23.3 81.3 Girls 160.1 21.1 82.0 Ages 15 to 19 [30] Sex Incidence Mortality Observed Survival % Boys 237.7 34.5 80.0 Girls 235.5 24.7 85.4 Note: Incidence and mortality rates are per 1,000,000 and age-adjusted to the 2000 US standard population. ... World Health Organization Initiative for Vaccine Research . Retrieved January 28, 2013 . ^ Moormann AM, Snider CJ, Chelimo K (October 2011).RARG, TAGLN, TP53, PROX1-AS1, GLRA3, PTPRN2, MYCN, KMT5B, ALK, RUNX1, CDKN2A, SMARCB1, RB1, ETV6, MRC1, BRCA2, H3P10, CD6, OPN1SW, AMH, PMS2, EWSR1, FOXO1, BUB1B, CBR3, MSH6, MIR132, ESR1, PIK3CD, PIK3CB, PIK3CA, PPIG, MLH1, PIK3CG, BRCA1, PALB2, CDKN2B, CRYZ, FGF23, TYRO3, H4C5, H4C2, THBS1, H4C8, H4C15, H4C3, H4C11, H4C12, H4C6, TNF, TSHR, VEGFA, CDKN2B-AS1, H4C4, EZR, H4C1, GFI1B, H3P9, VIM, H4C9, ARID1A, H4C14, BEST1, YWHAE, CXADRP1, H4C13, SNURF, MIR34A, UGT1A6, VANGL2, ARID1B, SLC28A3, FTO, RASSF4, BRIP1, BRSK1, MCM8, KDM2B, H4-16, MTFMT, LYPD4, GPC2, ASPG, MIR204, SLC12A9, DGCR8, ABCC3, SCARA3, BANF1, ARHGEF7, LIN28B, ARTN, EXO1, GDF15, NR1I3, CCS, TRIM13, TH, SUB1, MORF4, RASSF1, HPGDS, NXT1, MRPL28, AGRP, TFRC, S1PR1, ERBB2, ERCC1, ESR2, F3, FHIT, FOXO3, B4GALNT1, GFI1, GH1, GJB2, GNAS, GPR35, HMOX1, HTC2, IFNG, EPAS1, DPP4, AURKA, NQO1, APEX1, ARR3, ARSA, CCND1, CASR, CBR1, CD19, CD38, CDKN1A, CHEK1, CXADR, CYP3A4, DAXX, DCN, ACE, IGF1, IGF1R, IGF2R, JAK3, PRKAR1A, PTPN11, PVT1, RAD9A, BRD2, SCT, SHH, SIX1, SMARCA1, SMN1, SMN2, SNRPN, SOS1, SPG7, STAT3, PPARG, SLC26A4, PCSK1, KMT2A, KLRD1, STMN1, LEPR, LMNB1, MDM2, MDM4, MPI, PAX3, ABCC1, MSH2, MSH3, MTHFR, MUTYH, CNTN3, RENBP

-
Omphalocele
Wikipedia
"Prenatal and postnatal management of omphalocele" . Prenatal Diagnosis . 28 (7): 626. doi : 10.1002/pd.2008 . ... "Prenatal and postnatal management of omphalocele". Prenatal Diagnosis . 28 (7): 626–32. doi : 10.1002/pd.2008 .CHRNA7, PCSK5, LRP1, CDKN1C, FLNA, IGF2, KCNQ1OT1, PIGN, MKS1, RAB23, MBTPS2, DACT1, IFT81, LMOD1, ACTG2, GRIP1, WDR60, SPECC1L, CD96, SEMA3E, TP63, YWHAE, ADNP, CHD7, THRA, TTC7A, BHLHA9, FREM2, H19, HYLS1, FREM1, CEP120, TWIST2, NEK9, WDR34, FRAS1, DYNC2H1, PORCN, NXN, IFT80, WDR35, TSHB, H19-ICR, ISL1, HOXD13, KCNQ1, LBR, LRP2, MMP2, GPC3, MMP14, GJB2, MYH11, MYLK, NFIX, PAFAH1B1, FLNB, FGFR1, PLOD1, GPC4, COL11A2, MASP1, COL11A1, CHUK, HIC1, TCN2, AFP, MTHFR, EYA2, IH, BHMT, SLC19A1, CCL21, MAPK1, EPHB2, EYA1, WDR20, GYPE, GDF11, PITX2, SDS, CD320, GYPA, GYPB, TGM1

-
Mitochondrial Complex I Deficiency, Nuclear Type 1
Omim
In a study of 157 patients with respiratory chain defects, von Kleist-Retzow et al. (1998) found complex I deficiency in 33% and combined complex I and IV deficiency in another 28%. The main clinical features in this series were truncal hypotonia (36%), antenatal (20%) and postnatal (31%) growth retardation, cardiomyopathy (24%), encephalopathy (20%), and liver failure (20%). ... A high rate of parental consanguinity was observed in complex IV (20%) and complex I+IV (28%) deficiencies. Loeffen et al. (2000) retrospectively examined clinical and biochemical characteristics of 27 patients, all of whom presented in infancy and young childhood with isolated enzymatic complex I deficiency established in cultured skin fibroblasts; common pathogenic mtDNA point mutations and major rearrangements were absent.NDUFS4, NDUFS6, NDUFA1, NDUFV1, NDUFS1, NUBPL, NDUFS2, NDUFAF5, NDUFAF2, NDUFS3, FOXRED1, NDUFB3, NDUFS7, NDUFAF1, NDUFS8, NDUFV2, NDUFA11, NDUFB9, NDUFAF3, NDUFAF4, NDUFB11, ND2, NDUFB8, NDUFA6, ELAC2, TIMMDC1, NDUFB10, ND3, ND1, TMEM126B, AIFM1, SLC25A10, NDUFA13, ND4, RPS6KB1, GDAP1, SIRT3, NDUFAF6, FMN1, STXBP1
-
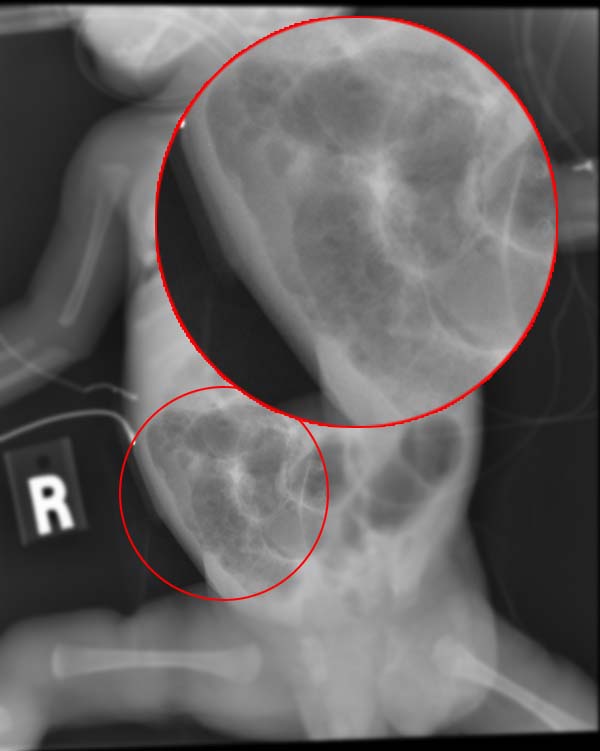
Necrotizing Enterocolitis
Wikipedia
Rates of death were almost three times higher for the black populations than for the white populations. [27] Overall, about 70-80% of infants who develop NEC survive. [28] Medical management of NEC shows an increased chance of survival compared to surgical management. [28] Despite a significant mortality risk, long-term prognosis for infants undergoing NEC surgery is improving, with survival rates of 70–80%.TLR4, HBEGF, NOS3, ACADM, SNAI3, CRYL1, PARK7, AKR1A1, SORD, SOD2, OAT, LTF, LGALS2, HSPA9, ENO1, NOS2, ALDH2, EGF, TNF, TLR2, IKBKG, CD14, TLR9, IL18, TLR1, TLR6, CD40, CD40LG, MUC2, TLR7, TLR5, TJP1, IL10RB, IL10, PTAFR, IL1B, PARP1, TUBB, TLR3, BANP, ADCY4, VEGFA, MBL2, IL6, MIF, HGF, LGR5, NDUFS7, SIGIRR, NOD2, EPO, DEFB4B, MIR431, VIP, MIR1290, CLDN2, NR1I2, ALB, ARRB2, CCR9, DEFB103A, IL22, IL23A, TOLLIP, ATG16L1, DEFB103B, IL33, IL23R, CTNNB1, SLC10A2, ASL, IL17A, DEFB4A, SLC25A10, EGFR, CRP, FCGRT, FCN1, GLRX, HMGB1, FOXA1, CRHR2, IGF1, IL4, IL4R, IL11, IRF5, THBD, CRHR1, CPS1, SMAD7, MAP3K5, MMP3, COX2, ACTA1, PPARG, MAPK8, PTGS2, REN, DEFA6, CAV1, TGFB2, MTCO2P12
-
Congenital Disorders Of N-Linked Glycosylation And Multiple Pathway Overview
Gene_reviews
For some individuals with MPI-CDG ( CDG-Ib ), heparin therapy can be an alternative to mannose in the treatment of the enteropathy [de Lonlay & Seta 2009]. A woman age 28 years with MPI-CDG ( CDG-Ib ) developed progressive liver fibrosis despite mannose treatment and heparin therapy and had a successful liver transplant with resolution of her symptoms for at least two years post transplant [Janssen et al 2014].PMM2, SRD5A3, MPI, SLC35A2, MOGS, DPM1, DOLK, MPDU1, SLC35A1, FCSK, ALG1, MAGT1, SRD5A3-AS1, TMEM165, PGM1, COG8, ALG6, COG6, ALG3, ALG9, PGM3, ALG8, DPAGT1, MAN1B1, COG7, COG5, APOC3, MGAT2, ALG12, DPM3, SLC35C1, SERPINA1, LEMD3, ALPI, ALPP, ATP6V0A2, ALG11, SLC39A8, STT3B, MAN1A1, GOLPH3, CD47, STT3A, IAPP, DHDDS, RFT1, ALG2, PGAP3, NUS1P3, COG2, GFM1, SLC35A3, POFUT2, CCDC115, NUS1, PGAP1, ALG13, CSGALNACT1, TRAPPC11, GRIN3A, AGA, RXYLT1, IGF1, GPI, B4GALT1, FUT8, FXN, F11, SLC26A3, DLD, DDOST, DCN, DAG1, SERPINA6, BGN, BCHE, ATP6AP1, SERPINC1, APRT, APOB, HP, IGFBP5, MGAM, ITGB2, ST3GAL5, DPM2, TUSC3, AKR1B1, TF, SSR4, SSR3, ST3GAL3, SI, RNASE4, RAB1A, PYCR1, ATXN3, MFAP1, LAMP2, LAMP1, LAD1, PGR-AS1
-
Arthrogryposis, Renal Dysfunction, And Cholestasis 1
Omim
Germline VPS33B mutations were present in 28 of 35 families (48 of 62 individuals); heterozygosity was found in the VPS33B locus in some cases of ARCS, suggesting the possibility of a second ARC syndrome gene.












