Colobomatous microphthalmia-obesity-hypogenitalism-intellectual disability syndrome is a rare, genetic, syndromic microphthalmia disorder characterized by bilateral, usually asymmetrical, microphthalmia associated typically with a unilateral coloboma, truncal obesity, borderline to mild intellectual disability, hypogenitalism and, more variably, nystagmus, cataracts and developmental delay.
Verloes et al. (1997) suggested the existence of an autosomal dominant coloboma-obesity-hypogenitalism-mental retardation syndrome. (See the Biemond syndrome II (210350) for an autosomal recessive form.)
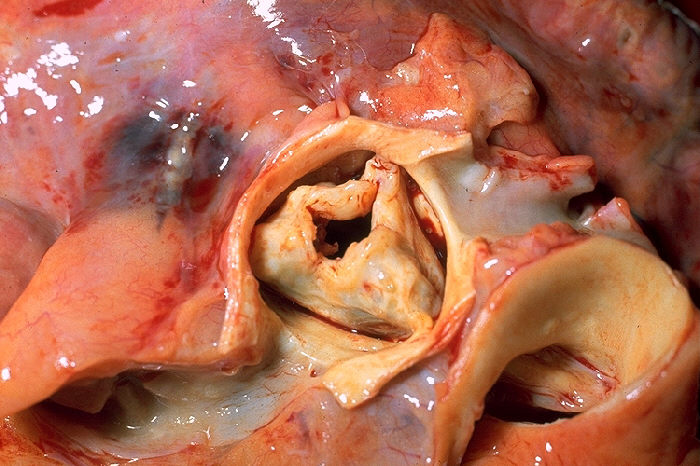
Cardiac anomalies-heterotaxy syndrome is characterised by non-compaction of the ventricular myocardium, bradycardia, pulmonary valve stenosis, and secundum atrial septal defect. Laterality sequence anomalies are also present. So far, the syndrome has been described in nine members from three generations of the same family.
Growing teratoma syndrome Growing teratoma syndrome is a rare complication of teratoma that can occur when an immature ovarian germ cell teratoma is treated by chotherapy. [1] References [ edit ] ^ "Ovarian Cancer" .
Condition in which severely overweight people fail to breathe rapidly or deeply enough Obesity hypoventilation syndrome Other names Pickwickian syndrome Obesity hypoventilation syndrome often improves with positive airway pressure treatment administered overnight by a machine such as this device Specialty Respirology Obesity hypoventilation syndrome ( OHS ) is a condition in which severely overweight people fail to breathe rapidly or deeply enough , resulting in low oxygen levels and high blood carbon dioxide (CO 2 ) levels. ... Sleep-related hypoventilation/hypoxemic syndromes. Chest. 2007;131(6):1936-48. ^ American Academy of Sleep Medicine. ... "Recent advances in obesity hypoventilation syndrome" . Chest . 132 (4): 1322–36. doi : 10.1378/chest.07-0027 . ... Treatment and prognosis of the obesity hypoventilation syndrome. UpToDate Aug 6, 2019. https://www.uptodate.com/contents/treatment-and-prognosis-of-the-obesity-hypoventilation-syndrome ^ Burwell CS, Robin ED, Whaley RD, Bicklemann AG (1956). ... Mr. Dickens' "Pickwickian" Fat Boy Syndrome" . Obesity Research . 2 (4): 380–383. doi : 10.1002/j.1550-8528.1994.tb00079.x .
Falsetti et al. (1964) described brother and sister with a syndrome characterized by obesity, cyanosis, somnolence, muscular twitching, and periodic breathing.
Papillon-Lefevre syndrome Other names Palmoplantar keratoderma with periodontitis Papillon–Lefèvre syndrome has an autosomal recessive pattern of inheritance . ... "Dermatalogical and oral findings in a cohort of 47 patients with Papillon-Lefevre syndrome". J Am Acad Dermatol . 48 (3): 345–351. doi : 10.1067/mjd.2003.197 . ... "Description of two new cathepsin C gene mutations in patients with Papillon-Lefèvre syndrome". J. Periodontol . 77 (2): 233–237. doi : 10.1902/jop.2006.050124 . ... PMID 16332247 . ^ Online Mendelian Inheritance in Man (OMIM): 602365 ^ a b c d e "Papillon Lefèvre Syndrome - NORD (National Organization for Rare Disorders)" . ... No. 6: 14. ^ Alchab, Dr. Izdihar. "Papillon–Lefèvre syndrome treatment with partial bone graft technique" .
A number sign (#) is used with this entry because Papillon-Lefevre syndrome (PALS) is caused by homozygous or compound heterozygous mutation in the cathepsin C gene (CTSC, or DPPI; 602365) on chromosome 11q14. Mutations in the CTSC gene also cause Haim-Munk syndrome (HMS; 245010) and aggressive periodontitis-1 (170650). ... Gorlin et al. (1964) suggested that calcification of the dura mater is a third component of the syndrome. Nazzaro et al. (1988) reported 4 sibs with Papillon-Lefevre syndrome, ranging in age from 2 to 11 years. ... No significant correlation could be demonstrated between the level of periodontal infection and severity of skin affections, supporting the concept that these 2 major components of Papillon-Lefevre syndrome are unrelated to each other. Almuneef et al. (2003) described a Saudi male with pyogenic liver abscesses, born to consanguineous parents, who was found to have Papillon-Lefevre syndrome. ... Gorlin et al. (1976) had suggested that PLS and Haim-Munk syndrome (HMS; 245010) were clinical variants.
Papillon-Lefèvre syndrome (PLS) is a rare ectodermal dysplasia characterized by palmoplantar keratoderma associated with early-onset periodontitis. ... Differential diagnosis Differential diagnosis includes two rare disorders that are allelic variants of PLS, Haim-Munk syndrome (see this term) and prepubertal/aggressive periodontitis. Other diseases with similar dermatologic features include localized epidermolytic palmoplantar keratoderma (Vörner), mal de Meleda, Howel-Evans syndrome, transgrediens et progrediens palmoplantar keratoderma (Greither's disease) (see these terms), and keratosis punctata.
Not to be confused with Bronze baby syndrome . Gray baby syndrome Specialty Pediatrics Diagnostic method proper history taking, monitoring blood level of the drug. Gray baby syndrome (also termed Gray or Grey syndrome ) is a rare but serious side effect that occurs in newborn infants (especially premature babies) following the accumulation of antibiotic chloramphenicol . [1] Contents 1 Signs and symptoms 2 Pathophysiology 3 Diagnosis 4 Prevention 5 Treatment 6 References 7 Further reading 8 External links Signs and symptoms [ edit ] Toxic levels of chloramphenicol after 2–9 days result in: loss of appetite, vomiting, ashen gray color of the skin, Hypotension (low blood pressure), Cyanosis (blue discolouration of lips and skin), Hypothermia , Cardiovascular collapse , hypotonia, abdominal distension, irregular respiration and increased blood lactate. [2] Pathophysiology [ edit ] Two pathophysiologic mechanisms are thought to play a role in the development of gray baby syndrome after exposure to the anti-microbial drug chloramphenicol. ... The cause of gray baby syndrome comes from the mother's use of an antibiotic, chloramphenicol, during pregnancy or breastfeeding. ... PMID 15249753 . ^ a b c Ed, Cummings; Ma, Edens (2020). "Gray Baby Syndrome" . PubMed . PMID 28846297 . Retrieved 2020-07-27 . ^ Brunton, Laurence L.; Lazo, John S.; Parker, Keith, eds. (2005). ... Further reading [ edit ] Krasinski, K; Perkin, R; Rutledge, J (1 September 1982). "Gray Baby Syndrome Revisited". Clinical Pediatrics . 21 (9): 571–572. doi : 10.1177/000992288202100910 .
Heyde's syndrome A stenotic aortic valve Specialty Cardiology , general surgery Heyde's syndrome is a syndrome of gastrointestinal bleeding from angiodysplasia in the presence of aortic stenosis . [1] [2] It is named after Edward C. ... Retrieved 9 July 2015 . ^ a b c Massyn MW, Khan SA (2009). "Heyde syndrome: a common diagnosis in older patients with severe aortic stenosis" . ... "From clinical observation to mechanism--Heyde's syndrome". The New England Journal of Medicine . 367 (20): 1954–6. doi : 10.1056/NEJMcibr1205363 . ... "An epidemiological study of Heyde's syndrome: an association between aortic stenosis and gastrointestinal bleeding". ... "Recurrent Gastro-intestinal Bleeding Unfolded as Heyde's Syndrome". The Indian Journal of Pediatrics . 85 (7): 589–590. doi : 10.1007/s12098-017-2587-7 .
"Construction and validation of a questionnaire distinguishing a chronic abdominal wall pain syndrome from irritable bowel syndrome" . ... "Abdominal cutaneous nerve entrapment syndrome". Surgery . 71 (1): 118–24. PMID 4332389 . ^ Scheltinga, M. ... "Management of anterior cutaneous nerve entrapment syndrome in a cohort of 139 patients". Annals of Surgery . 254 (6): 1054–8. doi : 10.1097/SLA.0b013e31822d78b8 . ... "Anterior cutaneous nerve entrapment syndrome (ACNES): The forgotten diagnosis". ... "Surgical options after a failed neurectomy in anterior cutaneous nerve entrapment syndrome". World Journal of Surgery . 38 (12): 3105–11. doi : 10.1007/s00268-014-2737-2 .
A chronic neuropathic pain syndrome of the abdominal wall caused by entrapment of anterior cutaneous branches of 7 to 12th intercostal nerves along the lateral border of the anterior rectus abdominis fascia causing severe pain and tenderness of the involved dermatome.
This syndrome is characterized by the association of bilateral microtia with severe to profound hearing impairment, and cleft palate. Epidemiology It has been described in four individuals from a consanguineous Iranian family. Etiology The syndrome is caused by point mutations in the HOXA2 gene, a gene that has already been shown to be involved in development of the auditory system in mice.
A number sign (#) is used with this entry because of evidence that microtia, hearing impairment, and cleft palate is caused by homozygous mutation in the HOXA2 gene (604685) on chromosome 7p15. One such family has been reported. There is also evidence that microtia with or without hearing impairment is caused by heterozygous mutation in the HOXA2 gene. Clinical Features Alasti et al. (2008) examined 3 of 4 affected members of a consanguineous Iranian family who had bilateral microtia, mixed symmetric severe to profound hearing impairment, and partial cleft palate. The helix, tragus, and antitragus were seen in all patients. On otoscopy, the external auditory canal was short and severely narrowed bilaterally, appearing almost stenotic; the disease was categorized as microtia grade II. A partial cleft palate was found in all 3 patients, 1 of whom also had right-sided facial paresis.
Trichodysplasia-xeroderma syndrome is an extremely rare, syndromic hair shaft anomaly characterized by sparse, coarse, brittle, excessively dry and slow-growing scalp hair, sparse axillary and pubic hair, sparse or absent eyelashes and eyebrows and dry skin.
Pinheiro and Freire-Maia (1987) described a distinctive disorder in many members of 6 generations of a family. The disorder combined various degrees of trichodysplasia and xeroderma. Trichodysplasia was a general term they used for all types of hair disturbances from alopecia and hypotrichosis to structural changes such as pili torti and trichorrhexis nodosa. The proband, a 30-year-old man, was healthy but had sparse, coarse, brittle, slow-growing and excessively dry scalp hair. Eyebrows were irregularly sparse and eyelashes were scanty and short.
Microcephaly-brain defect-spasticity-hypernatremia syndrome is a rare congenital genetic syndrome with a central nervous system malformation as a major feature characterized by microcephaly, hypertonia, developmental delay and cognitive impairment, swallowing difficulty, hypernatremia, and hypoplasia of the frontal parts and fusion of the lateral ventricles on brain MRI.
Distal 16p11.2 microdeletion syndrome is a rare chromosomal anomaly syndrome resulting from the partial deletion of the short arm of chromosome 16 with a highly variable phenotype typically characterized by developmental delay, mild intellectual disability and autism spectrum disorder.
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome (chr16:28.73-28.95 Mb). Description The deletion of a 220-kb region on chromosome 16p11.2 encompassing approximately 9 genes, including the SH2B1 gene (608937), is associated with a highly penetrant form of isolated severe early-onset obesity as well as obesity with developmental delay (summary by Bachmann-Gagescu et al., 2010). An extended 1.7-Mb deletion of chromosome 16p11.2 containing both the 220-kb region and the proximal 593-kb region associated autism (see 611913) has been reported in 2 patients with a syndrome of autism, mental retardation, and obesity and in 2 patients with pervasive developmental disorder, auditory processing difficulties, and attention deficit-hyperactivity disorder but not obesity.
Fetal akinesia-cerebral and retinal hemorrhage syndrome is a rare, lethal, congenital myopathy syndrome characterized by decreased fetal movements and polyhydraminos in utero and the presence of akinesia, severe hypotonia with respiratory insufficiency, absent reflexes, joint contractures, skeletal abnormalities with thin ribs and bones, intracranial and retinal hemorrhages and decreased birth weight in the neonate.
A number sign (#) is used with this entry because of evidence that lethal congenital contracture syndrome-5 (LCCS5) is caused by homozygous mutation in the DNM2 gene (602378) on chromosome 19p13. ... Clinical Features Koutsopoulos et al. (2013) reported 3 sibs, born of consanguineous Pakistani parents, with a lethal congenital neuromuscular syndrome. All exhibited decreased fetal movements, polyhydramnios, and decreased birth weight. ... Inheritance The transmission pattern of a lethal congenital contracture syndrome in the family reported by Koutsopoulos et al. (2013) was consistent with autosomal recessive inheritance. Molecular Genetics In 3 sibs, born of consanguineous Pakistani parents, with a lethal congenital contracture syndrome, Koutsopoulos et al. (2013) identified a homozygous missense mutation in the DNM2 gene (F379V; 602378.0013).
X-linked lethal multiple pterygium syndrome is a rare, genetic, developmental defect during embryogenesis characterized by the typical lethal multiple pterygium syndrome presentation (comprising of multiple pterygia, severe arthrogryposis, cleft palate, cystic hygromata and/or fetal hydrops, skeletal abnormalities and fetal death in the 2nd or 3rd trimester) with an X-linked pattern of inheritance.
Carnevale et al. (1973) observed a family with 7 cases of pterygium syndrome in 3 generations and suggested X-linked dominant inheritance because father-to-son transmission did not occur, and all 4 daughters but none of 4 sons of an affected male were affected. ... Tolmie et al. (1987) described a prenatal lethal multiple pterygium syndrome occurring in 2 male sibs and a first cousin once removed connected through presumptively carrier females. Meyer-Cohen et al. (1999) described 4 male fetuses in 1 sibship with healthy nonconsanguineous parents and raised the question of an X-linked recessive subtype of lethal pterygium syndrome. In a review of the literature, the family reported by Tolmie et al. (1987) was the only one that strongly supported X-linked inheritance.
A rare neurologic disease characterized by the presence of Duane retraction syndrome (i. e. a congenital cranial dysinnervation disorder with unilateral or bilateral limitation of abduction and/or adduction of the eye, as well as globe retraction and palpebral fissure narrowing on attempted adduction) in combination with congenital unilateral or bilateral hearing loss. The sidedness of hearing loss corresponds to the sidedness of the retraction syndrome.
Orofaciodigital syndrome type 11 is an extremely rare, sporadic form of Orofaciodigital syndrome (OFDS; see this term) with only a few reported cases, and characterized by facial (blepharophimosis, bulbous nasal tip, broad nasal bridge, downslanting palpebral fissures and low set ears) and skeletal (post-axial polydactyly and fusion of vertebrae) malformations along with severe intellectual disability, deafness and congenital heart defects.
Clinical Features Gabrielli et al. (1994) reported a boy with an oral-facial-digital syndrome associated with skeletal anomalies and severe psychomotor delay. ... Gabrielli et al. (1994) suggested that this was a novel OFD syndrome associated with vertebral and craniofacial anomalies. Ferrero et al. (2002) described a girl with an OFD syndrome associated with a nasopharyngeal hairy polyp, partial synostosis between the atlas and the base of the occipital bone, multiple proximal-cervical and distal-thoracic vertebral clefts, kyphoscoliosis, empty sella turcica, and mental retardation. ... Obregon and Barreiro (2003) suggested that the patient had Gabrielli syndrome. They noted that she did not have cleft palate and oral frenula, but they considered facial anomalies, cardiac defects, skeletal abnormalities, mental retardation, and hearing loss to be the cardinal manifestations of the syndrome.
Karsch-Neugebauer syndrome is a rare syndrome characterized by split-hand and split-foot deformity and ocular abnormalities, mainly a congenital nystagmus.
Pilarski et al. (1985) reported this syndrome in a mother and 3 of her children. ... Wong et al. (1998) reported on 2 sisters with Karsch-Neugebauer syndrome (KNS) comprising split foot and split hand anomalies in association with congenital nystagmus.
Split hand split foot nystagmus is a rare congenital syndrome characterized by split hand and split foot deformity and eye abnormalities, especially nystagmus .
Pitt-Hopkins-like syndrome is a rare, genetic, syndromic intellectual disability disorder characterized by severe intellectual disability, lack of speech with normal, or mildly delayed, motor development, episodic breathing abnormalities, early-onset seizures and facial dysmorphism which only includes a wide mouth.
A number sign (#) is used with this entry because of evidence that Pitt-Hopkins-like syndrome-1 (PTHSL1) is caused by homozygous or compound heterozygous mutation in the CNTNAP2 gene (604569) on chromosome 7q35-q36. ... Clinical Features Strauss et al. (2006) described a clinical and neuropathologic phenotype, which they called cortical dysplasia-focal epilepsy syndrome (CDFES), in 9 children in the Old Order Amish of Lancaster, Pennsylvania. ... The long-term outcome was poor, with patients fully dependent on others for daily living. Pitt-Hopkins-Like Syndrome 1 Orrico et al. (2001) described 2 sibs, a brother and sister, with severe mental retardation and multiple congenital anomalies including coarse facial features, short stature, seizures, hypertrichosis, short great toes, and overbreathing. ... Mapping Strauss et al. (2006) mapped the CDFE syndrome to 7q36 by demonstration of a large block of putative autozygosity. ... The phenotype overlapped that of Pitt-Hopkins syndrome and is designated here as Pitt-Hopkins-like syndrome-1.
A rare genetic epilepsy characterized by relatively large head circumference or macrocephaly, diminished or absent deep-tendon reflexes and mild gross motor delay in infancy, followed by intractable focal seizures with language regression, behavioral abnormalities (hyperactivity, attention deficit, aggressive/autoaggressive behavior, autistic features) and intellectual disability later in life.