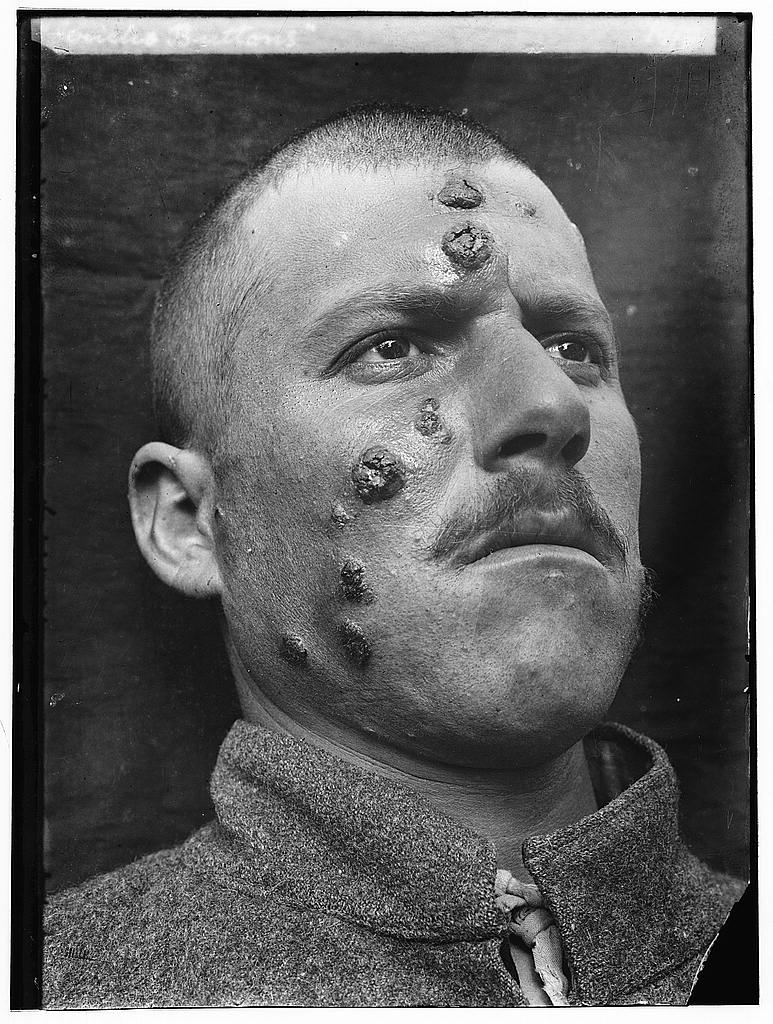
Autoantibodies and cold agglutinins may be present and 10% of CMML is DCT positive. [7] [9] Bone marrow aspirates will display hypercellularity with increased counts of granulocytic and monocytic cells. [1] Bone marrow core biopsies may show a predominance of myelocytic and monocytic cells, abnormal localisation of immature precursors and dysplastic megakaryocytes . [1] Monocytic nodules are a common feature in biopsies. [16] The phenotypical characteristics of CMML are; CD11b , CD11c , CD14 , CD33 , CD45 and CD64 seen in 100% of cases; CD13 found in 95% of cases; CD4 found in 76% of cases; HLA-DR found in 71% of cases; CD56 found in 53% of cases; CD2 found in 34% of cases; CD16 found in 29% of cases; CD10 found in 28% of cases; CD23 and CD7 found in 9% of cases; and CD117 found in 5% of cases. [17] Classification [ edit ] Haematopoiesis. ... However, due to the late age of onset and presence of other illnesses, this form of treatment is often not possible. [5] [28] Epidemiology [ edit ] There have been few individual epidemiological studies of CMML, due to the difficulty in the disease classification.
A rare myelodysplastic/myeloproliferative neoplasm characterized by a spectrum of clinical, hematological, and morphological features, ranging from predominantly myelodysplastic to mainly myeloproliferative in nature. Infiltration of the liver, spleen, lymph nodes, and other organs is common. Persistent peripheral blood monocytosis with monocytes accounting for more than 10% of leukocytes is the hallmark of the condition. Blasts constitute less than 20% of the cells in the peripheral blood and bone marrow. Other abnormalities are variable. Patients may present with constitutional symptoms, signs and symptoms of hematopoietic insufficiency, and hepatosplenomegaly.
During the process, the therapist can ask hypothetical questions in a form of therapeutic Socratic questioning . [28] This therapy has been mostly studied in patients with the persecutory type. ... Archived from the original on 2015-04-02 . Retrieved 2015-03-28 . CS1 maint: archived copy as title ( link ) ^ http://steinhardt.nyu.edu/opus/issues/2010/spring/shutter_island ^ Mammootty, Adoor; Asokan; Shobana; Bahadur (1987-10-01), Anantaram , retrieved 2017-02-02 ^ "Anantaram: After three years, another landmark from Adoor Gopalakrishnan" .
With an 800-microsecond TMS pulse and a 28 ms stimulus at 11 degrees per second, V5 is incapacitated for about 20–30 ms. ... PMID 15707904 . ^ a b c Zihl, J., ULM Munich (Max Planck Institute of Psychiatry), interviewed by R. Hamrick, Oct. 28, 2009. ^ a b c d e f Beckers G, Zeki S (February 1995).
Thus it is recommended that multiple strains of the bacteriophage be used in each application so the bacteria do not have a chance to develop resistance to any one strain. [28] Importance [ edit ] The disease was first identified in the western states of, California, Washington, Texas, Arizona and Idaho in the 1970s and initially led to substantial yield losses in those areas. [15] Erwinia caratovara subsp betavascularum was not discovered in Montana until 1998. ... Archived from the original on 23 September 2015 . Retrieved 28 September 2013 . ^ Smigocki, A C.
It does not survive in the environment for more than a few hours at room temperature (20–25 °C), but can survive for a few weeks in shady environments at temperatures slightly above freezing. [28] It, along with other labile viruses, can also persist longer in serum and tissue debris. [23] Despite extensive vaccination in many regions, it remains a major disease of dogs. [29] To prevent canine distemper, puppies should begin vaccination at 6-8 weeks of age and then continue getting the “booster shot” every 2-4 weeks until they are 16 weeks of age. ... HealthCommunities.com . 4 Nov 2014 [28 Feb 2001]. Archived from the original on 2014-12-20 .
Statistics from U.S. healthcare report 18.1% re-admittance rate within 30 days for patients who undergo SBO surgery. [27] More than 90% of patients also form adhesions after major abdominal surgery. [28] Common consequences of these adhesions include small-bowel obstruction, chronic abdominal pain, pelvic pain, and infertility. [28] Animals [ edit ] Main article: Impaction (animals) References [ edit ] ^ a b c d e f g h i j k l Gore RM, Silvers RI, Thakrar KH, Wenzke DR, Mehta UK, Newmark GM, Berlin JW (November 2015).
Overview Intestinal obstruction is a blockage that keeps food or liquid from passing through your small intestine or large intestine (colon). Causes of intestinal obstruction may include fibrous bands of tissue (adhesions) in the abdomen that form after surgery; hernias; colon cancer; certain medications; or strictures from an inflamed intestine caused by certain conditions, such as Crohn's disease or diverticulitis. Colon and small intestine The small intestine and colon are parts of your digestive tract, which processes the foods you eat. The intestines take nutrients from the foods. What isn't absorbed by the intestines continues along the digestive tract and is passed as stool during a bowel movement. Without treatment, the blocked parts of the intestine can die, leading to serious problems.
For example, in one study, despite treatment with high doses of sodium stibogluconate for 28 days, only 30% of patients remained disease-free at 12 months follow-up. [10] Even in those patients who achieve an apparent cure, as many as 19% will relapse. [11] Several drug combinations with immunomodulators have been tested, for example, a combination of pentoxifylline (inhibitor of TNF-α ) and a pentavalent antimonial at a high dose for 30 days in a small-scale (23 patients) randomised placebo-controlled study from Brazil achieved cure rates of 90% and reduced time to cure, [12] a result that should be interpreted cautiously in light of inherent limitations of small-scale studies. [13] In an earlier small-scale (12 patients) study, addition of imiquimod showed promising results [14] which need yet to be confirmed in larger trials. ... Of the dozens of plants examined, the three plants that attracted especially sand flies are the Ochradenus baccatus, Prosopis farcta , and Tamarix nilotica . [28] Outbreak in 2016 [ edit ] The Middle East, in 2016, seems to be experiencing an increase in the cutaneous leishmaniasis disease due to migrants fleeing the Islamic State of Iraq and the Levant .
Behaviour Research and Therapy . 42 (3): 315–28. doi : 10.1016/s0005-7967(03)00141-4 . ... "A review of acute stress disorder in DSM-5". Depression and Anxiety . 28 (9): 802–817. doi : 10.1002/da.20737 .
Severe, sudden metabolic acidosis is a common cause of mortality. [12] Estimates of the rate of genetic carriers in the Saguenay-Lac-Saint-Jean region range from 1 in 23 to 1 in 28; the number of children born with the disease has been estimated at 1 in 2063 to 1 in 2473 live births. ... "First 'three person baby' born using new method" . BBC News . Retrieved 2016-09-28 . ^ Leigh, D (1951). "Subacute Necrotizing Encephalomyelopathy in an Infant" .
A progressive neurological disease defined by specific neuropathological features associating brainstem and basal ganglia lesions. Epidemiology Its prevalence at birth has been estimated at approximately 1 in 36 000. Clinical description Typical onset of symptoms occurs before the age of 12 months but, in rare cases, the disease may manifest during adolescence or even early adulthood. Loss of motor milestones, hypotonia with poor head control, recurrent vomiting, and a movement disorder are common initial symptoms. Pyramidal and extrapyramidal signs, nystagmus, breathing disorders, ophthalmoplegia and peripheral neuropathy are often noted later.
Leigh syndrome is a rare, inherited neurodegenerative condition . It usually becomes apparent in infancy, often after a viral infection . Rarely, it begins in the teenage or adult years. Signs and symptoms usually progress rapidly. Early symptoms may include poor sucking ability; loss of head control and motor skills; loss of appetite; vomiting; and seizures. As the condition progresses, symptoms may include weakness and lack of muscle tone ; spasticity; movement disorders; cerebellar ataxia ; and peripheral neuropathy. Complications can lead to impairment of respiratory, heart and kidney function.
Leigh syndrome is a severe neurological disorder that usually becomes apparent in the first year of life. This condition is characterized by progressive loss of mental and movement abilities (psychomotor regression) and typically results in death within two to three years, usually due to respiratory failure. A small number of individuals do not develop symptoms until adulthood or have symptoms that worsen more slowly. The first signs of Leigh syndrome seen in infancy are usually vomiting, diarrhea, and difficulty swallowing (dysphagia), which disrupts eating. These problems often result in an inability to grow and gain weight at the expected rate (failure to thrive).
Treatment using botulinum toxin type B is comparable to type A, with an increased frequency of the side effect dry mouth. [10] [20] Common side effects include pain at the injection site (up to 28%), dysphagia due to the spread to adjacent muscles (11% to 40%), dry mouth (up to 33%), fatigue (up to 17%), and weakness of the injected or adjacent muscle (up to 56%). [16] A Cochrane review published in 2016 reported moderate-quality evidence that a single Botulinum toxin-B treatment session could improve cervical dystonia symptoms by 10% to 20%, although with an increased risk of dry mouth and swallowing difficulties. [21] Another Cochrane review published in 2020 for Botulinum toxin-A found similar results. [22] Deep brain stimulation [ edit ] Insertion of electrode during surgery Deep brain stimulation to the basal ganglia and thalamus has recently been used as a successful treatment for tremors of patients with Parkinson's disease . ... A significant reduction in pain and severity of dystonia as well as increased postural awareness and quality of life was found. [25] Epidemiology [ edit ] Spasmodic torticollis is one of the most common forms of dystonia seen in neurology clinics, occurring in approximately 0.390% of the United States population in 2007 (390 per 100,000). [3] Worldwide, it has been reported that the incidence rate of spasmodic torticollis is at least 1.2 per 100,000 person years, [26] and a prevalence rate of 57 per 1 million. [27] The exact prevalence of the disorder is not known; several family and population studies show that as many as 25% of cervical dystonia patients have relatives that are undiagnosed. [28] [29] Studies have shown that spasmodic torticollis is not diagnosed immediately; many patients are diagnosed well after a year of seeking medical attention. [1] A survey of 59 patients diagnosed with spasmodic torticollis show that 43% of the patients visited at least four physicians before the diagnosis was made. [30] There is a higher prevalence of spasmodic torticollis in females; females are 1.5 times more likely to develop spasmodic torticollis than males.
Overview Cervical dystonia, also called spasmodic torticollis, is a painful condition in which your neck muscles contract involuntarily, causing your head to twist or turn to one side. Cervical dystonia can also cause your head to uncontrollably tilt forward or backward. A rare disorder that can occur at any age, cervical dystonia most often occurs in middle-aged people, women more than men. Symptoms generally begin gradually and then reach a point where they don't get substantially worse. There is no cure for cervical dystonia. The disorder sometimes resolves without treatment, but sustained remissions are uncommon.
Cervical dystonia is a neurological condition characterized by excessive pulling of the muscles of the neck and shoulder resulting in abnormal movements of the head (dystonia). Most commonly, the head turns to one side or the other. Tilting sideways, or to the back or front may also occur. The turning or tilting movements may be accompanied by shaking movement ( tremor ) and/or soreness of the muscles of the neck and shoulders. Cervical dystonia can occur at any age, but most cases occur in middle age. It often begins slowly and usually reaches a plateau over a few months or years.
., Wayanad (2013) and Malappuram districts of Kerala (2014), North Goa district of Goa state (2015), and Sindhudurg district of Maharashtra (2016). [27] Serological evidence for KFD [ edit ] There are reported serological evidence for KFD detected in humans in other parts of India, namely Kutch and Saurashtra regions of Gujarat state, Kingaon and Parbatpur of West Bengal state. [28] A seroprevalence study in Andaman and Nicobar islands in 2002 revealed a high prevalence of hemagglutination inhibition (HI) antibodies against KFDV. [29] Epidemiology [ edit ] The disease has a fatality rate of 3-10%, and it affects 400-500 people annually. [10] [14] The disease was first noted at Kyasanur village near Sagar in Shivamogga district of Karnataka. ... "Ticks (Ixodidae) on birds migrating from Europe and Asia to Africa, 1959-61" . Bull. World Health Organ . 28 (2): 235–262. PMC 2554471 . PMID 13961632 . ^ Lewis, Michael (2002).
Kyasanura forest disease (KFD), caused by the KFD virus, is an arbovirus characterized by an initial fever, headache and myalgia that can progress to a hemorrhagic disease and that in some cases is followed by a second phase characterized by neurological manifestations.
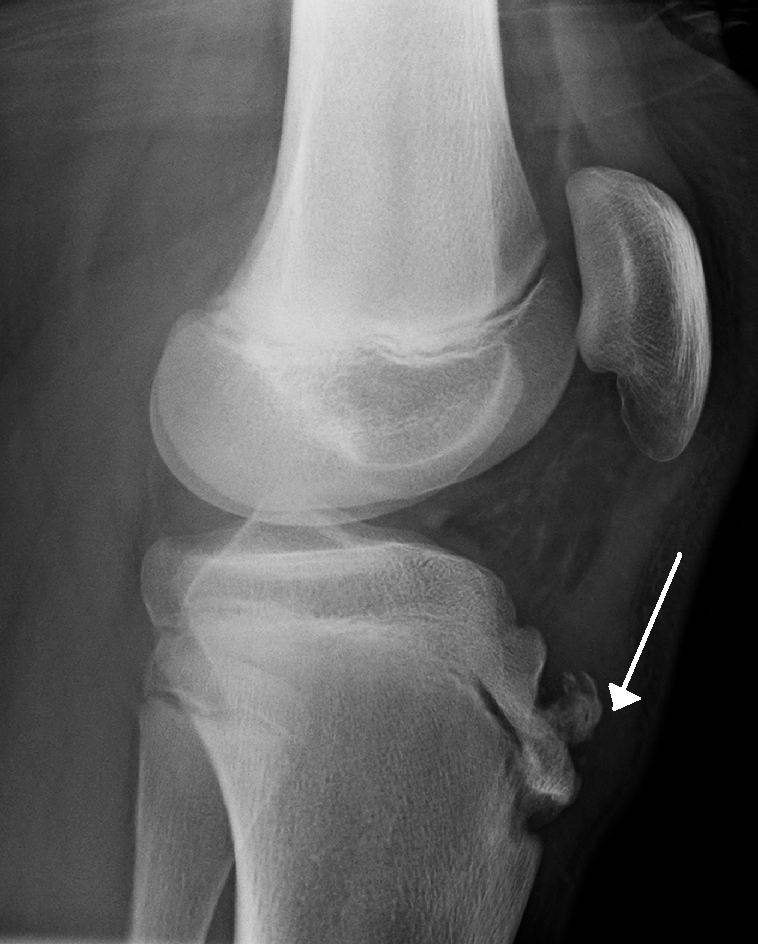
It has been suggested that difference is related to a greater participation by boys in sports and risk activities than by girls. [28] Osgood Schlatter’s disease resolves or becomes asymptomatic in majority of cases. ... "Osgood-schlatter disease: risk of a disease deemed banal" . Pan African Medical Journal . 28 : 56. doi : 10.11604/pamj.2017.28.56.13185 .
Osgood-Schlatter disease is a traction apophysitis of the anterior tibial tubercle described in active adolescents and characterized by gradual onset of pain and swelling of the anterior knee causing limping that usually disappears at the end of growth.
According to the doctors in one of these studies, the expected outcome for SMA syndrome treatment is generally considered to be excellent. [11] Epidemiology [ edit ] According to a 1956 study, only 0.3% of patients referred for an upper-gastrointestinal-tract barium studies fit this diagnosis, and is thus a rare disease . [27] Recognition of SMA syndrome as a distinct clinical entity is controversial, due in part to its possible confusion with a number of other conditions, [28] though it is now widely acknowledged. [1] However, unfamiliarity with this condition in the medical community coupled with its intermittent and nonspecific symptomatology probably results in its underdiagnosis. [29] As the syndrome involves a lack of essential fat, more than half of those diagnosed are underweight , sometimes to the point of sickliness and emaciation . ... Journal of Korean Medical Science . 28 (8): 1220–5. doi : 10.3346/jkms.2013.28.8.1220 .
Superior mesenteric artery syndrome (SMAS) is a digestive condition that occurs when the duodenum (the first part of the small intestine) is compressed between two arteries (the aorta and the superior mesenteric artery). This compression causes partial or complete blockage of the duodenum. Symptoms vary based on severity, but can be severely debilitating. Symptoms may include abdominal pain, fullness, nausea, vomiting, and/or weight loss. SMAS typically is due to loss of the mesenteric fat pad (fatty tissue that surrounds the superior mesenteric artery). The most common cause is significant weight loss caused by medical disorders, psychological disorders, or surgery.
The time between the infection and the start of the illness averages 28 days (ranging from 15 to 50 days), [6] and most recover fully within 2 months, although approximately 15% of sufferers may experience continuous or relapsing symptoms from six months to a year following initial diagnosis . [7] Hepatitis A [8] Marker Detection Time Description Significance Faecal HAV 2–4 weeks or 28 days – Early detection Ig M anti HAV 4–12 weeks Enzyme immunoassay for antibodies During acute Illness Ig G anti HAV 5 weeks–persistent Enzyme immunoassay for antibodies Old infection or reinfection Hepatitis B [ edit ] Main article: Hepatitis B Hepatitis B is caused by the hepatitis B virus, a hepadnavirus that can cause both acute and chronic hepatitis.
Kidney and cardiac functions need to be monitored closely as there are concerns about toxification . [28] [29] High dosage of chemotherapy may damage the bone marrow, in which autologous stem cell therapy is a recommended follow-up treatment. [7] Drugs [ edit ] Other common drugs used to treat mature T-cell lymphoma includes pralatrexate (fotolyn) , Brentuximab vedotin (Adcetris) and Romidepsin (Istodax) . [27] Pralatrexate is the first approved drug to treat lymphoma by the Food and Drug Administration (FDA) in 2009. [30] It is shown to reduce the size of lymphoma tumours. ... Seminars in Diagnostic Pathology . 28 (3): 202–213. doi : 10.1053/j.semdp.2011.03.003 .
An extremely rare, primary cutaneous T-cell lymphoma disorder characterized by solitary, or multifocal and diffuse, cutaneous lesions, ranging from tumor-like patches, plaques, papules, nodules, and/or erythroderma, located on any area of the body, which rapidly progress and may become ulcerated and/or infected. Systemic involvement may be associated.
No staging criteria exist for extraocular SGc, but the AJCC guidelines for nonmelanoma skin cancer or the eighth edition of the Union for International Cancer Control TNM staging system for skin carcinomas may be used. [2] [6] Sentinel lymph node biopsy [ edit ] Regional nodes are involved in as many as 10 to 28% of periocular SGc. Nodal involvement in extraocular SGc is less well studied. ... Data on the role of adjuvant radiation therapy in the treatment of SGc is scarce, however, and recurrence following adjuvant radiation therapy has been reported. [2] Prognosis [ edit ] Greater survival rates have been observed for ocular versus extraocular tumors and localized versus regional disease. [6] The observed survival rates at 5 and 10 years are 78.20 and 61.72%, respectively, while the relative survival rates at 5 and 10 years are 92.72 and 86.98%, respectively. [6] SGc is believed to spread through the blood and lymphatic system via three mechanisms: tumor growth, multifocal tumor proliferation and shedding of atypical epithelial cells that subsequently transplant in a distant site. [6] Due to difficulty in promptly diagnosing SGc, the rate of metastasis and recurrence is relatively high. [17] The rate of metastasis is approximately 4.4% for periocular SGc and 1.4% for extraocular SGc. [6] Periocular SGc frequently causes regional metastases resulting in a mortality rate of approximately 22%. [9] Periocular SGc most commonly metastasizes to regional lymph nodes and rarely the lungs, liver, brain, or bone. [5] Regional nodes are involved in as many as 10 to 28% of periocular SGc. Nodal involvement in extraocular SGc is less well studied. [6] At the time of diagnosis nearly 25% of tumors will metastasize.
IPLEX's manufacturing company, Insmed, after selling its protein production facility, can no longer develop proteins, thus can no longer manufacture IPLEX as of a statement released in July 2009. [24] Prognosis [ edit ] Cancer and Diabetes [ edit ] It has been reported that people with LS in Ecuador are resistant to cancer and diabetes and are somewhat protected against aging. [25] [26] [27] This is consistent with findings in mice with a defective growth hormone receptor gene. [20] Among the approximately 100 individuals in this population, there were no reported cases of diabetes and one case of cancer. [28] A 2019 study of individuals with isolated growth hormone deficiency (IGHD type 1B) in Itabaianinha County, Brazil demonstrated a phenotype consistent with Laron syndrome. [29] Researchers found that these humans had similarly extended healthspan , with resistance to cancer and attenuated effects of aging, but neither patients with LS nor IGHD experienced an increase in their overall lifespan. [29] Incidence [ edit ] The majority of reported cases of Laron syndrome have been in people with Semitic origins, almost all of them being Jews or assimilated descendants of Jews. [ citation needed ] Numerous Laron syndrome patients are found in Israel among the country's diverse Jewish population composed of Jews from around the world, as well as patients outside Israel originally from communities of the Jewish diaspora , such as Egypt and Iraq . ... Springer Science & Business Media. pp. 27–28. ISBN 978-3-642-11183-9 . Retrieved 10 November 2020 . ^ Shevah O, Kornreich L, Galatzer A, Laron Z (2005).
Laron syndrome is a condition that occurs when the body is unable to utilize growth hormone. It is primarily characterized by short stature. Other signs and symptoms vary but may include reduced muscle strength and endurance; hypoglycemia in infancy; delayed puberty; short limbs (arms and legs); and obesity . It is often caused by changes (mutations) in the GHR gene and is inherited in an autosomal recessive manner. Treatment is focused on improving growth and generally includes injections of insulin-like growth factor 1 (IGF-1).
A number sign (#) is used with this entry because of evidence that Laron syndrome, also known as growth hormone insensitivity syndrome, is caused by homozygous or compound heterozygous mutation in the growth hormone receptor gene (GHR; 600946) on chromosome 5p. Description Laron syndrome is an autosomal recessive disorder characterized by marked short stature that results from failure to generate insulin-like growth factor I (IGF1; 147440) in response to growth hormone (GH; 139250). GH levels are normal or increased. The disorder is caused by dysfunction of the growth hormone receptor. A Laron syndrome-like phenotype associated with immunodeficiency (245590) is caused by a postreceptor defect, i.e., mutation in the STAT5B gene (604260). Patients with mutations in the GHR gene that cause only partial insensitivity to growth hormone have a form of short stature (604271).
Laron syndrome is a rare form of short stature that results from the body's inability to use growth hormone , a substance produced by the brain's pituitary gland that helps promote growth. Affected individuals are close to normal size at birth, but they experience slow growth from early childhood that results in very short stature. If the condition is not treated, adult males typically reach a maximum height of about 4.5 feet; adult females may be just over 4 feet tall. Other features of untreated Laron syndrome include reduced muscle strength and endurance, low blood sugar levels (hypoglycemia) in infancy, small genitals and delayed puberty, hair that is thin and fragile, and dental abnormalities. Many affected individuals have a distinctive facial appearance, including a protruding forehead, a sunken bridge of the nose (saddle nose), and a blue tint to the whites of the eyes (blue sclerae).
Growth hormone insensitivity syndrome (GHIS) is a group of diseases characterized by marked short stature associated with normal or elevated growth hormone (GH) concentrations, which fail to respond to exogenous GH administration. GHIS comprises growth delay due to IGF-1 deficiency, growth delay due to IGF-1 resistance, Laron syndrome, short stature due to STAT5b deficiency and primary acid-labile subunit (ALS) deficiency (see these terms). Epidemiology A few cases of IGF-1 deficiency, IGF-1 resistance, STAT5B and ALS deficiencies, and more than 250 cases of Laron syndrome have been reported in the literature so far. Males and females are equally affected. Clinical description Intrauterine growth and birth size are usually subnormal. Postnatal growth is slowed. Facial dysmorphism is common and includes microcephaly, thin upper and everted lower lips and small chin.
Laron syndrome is a congenital disorder characterized by marked short stature associated with normal or high serum growth hormone (GH) and low serum insulin-like growth factor-1 (IGF-I) levels which fail to rise after exogenous GH administration. Epidemiology The disease has been described in more than 250 cases and is more frequent in Semitic or Mediterranean populations. Males and females are equally affected. Clinical description Intrauterine growth and birth size are usually normal. Postnatal growth is slowed and generally disproportional with delayed bone age; adult stature ranges from -3 to -12 SD. Motor development is delayed because of diminished muscle mass. Newborns often present with hypoglycemia and a micropenis.
Mutations in the gene OCLN on chromosome 5q13.2, which is thought to cause band-like calcification in the brain, have been discovered in affected individuals and categorized as BLCPMG which often associated with AGS. [12] [19] In most cases, except for IFIH1- and rare cases of TREX1- and ADAR1-related disease, these mutations follow an autosomal recessive inheritance pattern (and thus the parents of an affected child face a 1 in 4 risk of having a further child similarly affected at every conception). [ citation needed ] AGS can be divided into subtypes based on the gene in which the causative mutation occurs. [20] [21] A survey of 374 patients with an AGS diagnosis reported that the most frequent mutations occurred in RNASEH2B. [22] Type OMIM Gene Locus Frequency AGS1 225750 TREX1 3p21.31 23% (1% dominant) AGS2 610181 RNASEH2B 13q14.3 36% AGS3 610329 RNASEH2C 11q13.1 12% AGS4 610333 RNASEH2A 19p13.2 5% AGS5 612952 SAMHD1 20q11.23 13% AGS6 615010 ADAR 1q21.3 7% (1% dominant) AGS7 615846 IFIH1 2q24 3% (all dominant) AGS-associated mutations have been found to show incomplete penetrance in some cases, with children in the same family with the same mutations showing markedly different neurological and developmental outcomes. [22] Clinical features and disease course vary somewhat by genotype, with TREX1 associated with likely in utero onset and high mortality rate, [22] and RNASEH2B mutations associated with slightly milder neurological impairments, [23] lower interferon activity, and longer lifespan. [22] Pathology [ edit ] Type I interferon activity was originally described over 50 years ago as a soluble factor produced by cells treated with inactivated, non-replicating viruses that blocked subsequent infection with live virus. [24] [25] Although the rapid induction and amplification of the type I interferon system is highly adaptive in terms of virus eradication, aberrant stimulation or unregulated control of the system could lead to inappropriate and / or excessive interferon output. [26] Studies of the AGS-related proteins TREX1, the RNase H2 complex, SAMHD1 and ADAR1, suggest that an inappropriate accumulation of self-derived nucleic acids can induce type I interferon signaling. [27] [28] [29] The findings of IFIH1 mutations in the similar context implicates the aberrant sensing of nucleic acids as a cause of immune upregulation. [12] What is the source of the nucleic acid inducing the immune disturbance in AGS? ... PMID 1641084 . ^ "Proceedings of the International Meeting on Aicardi-Goutieres Syndrome Pavia, Italy, 28-29 May 2001". Eur J Paediatr Neurol . 6, Suppl A: A1–86. 2002. ^ a b Crow, YJ; et al. (2006).
Aicardi-Goutières syndrome is a disorder that mainly affects the brain, the immune system, and the skin. Most newborns with Aicardi-Goutières syndrome do not show any signs or symptoms of the disorder. However, about 20 percent are born with a combination of features that include an enlarged liver and spleen (hepatosplenomegaly), elevated blood levels of liver enzymes, a shortage of blood cells called platelets that are needed for normal blood clotting (thrombocytopenia), and neurological abnormalities. While this combination of signs and symptoms is typically associated with the immune system's response to a viral infection that is present at birth (congenital), no actual infection is found in these infants. For this reason, Aicardi-Goutières syndrome is sometimes referred to as a "mimic of congenital infection."
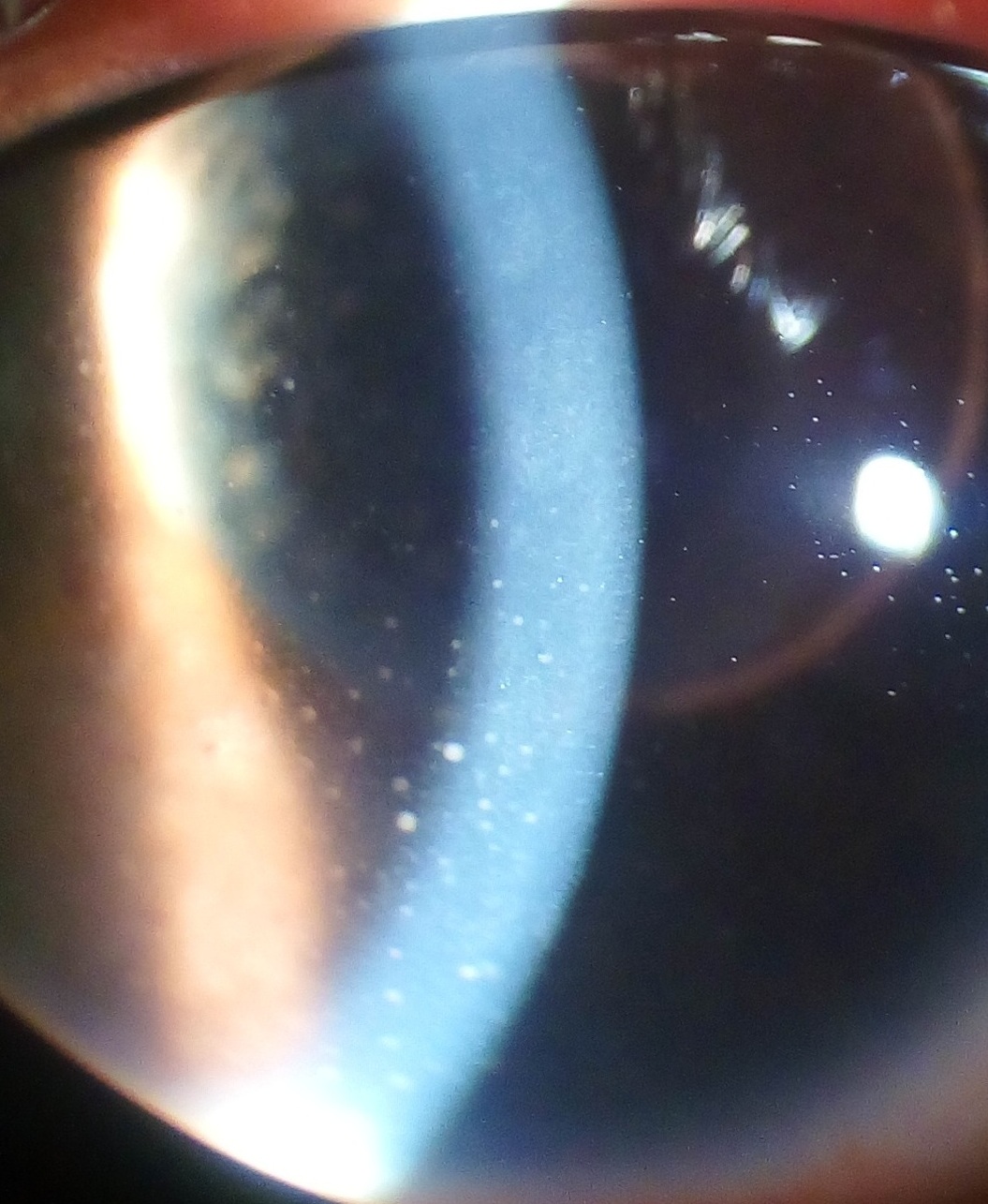
The anti-diabetic drug metformin is reported to inhibit the process that causes the inflammation in uveitis. [28] In the case of herpetic uveitis, anti-viral medications, such as valaciclovir or aciclovir , may be administered to treat the causative viral infection. [29] Prognosis [ edit ] The prognosis is generally good for those who receive prompt diagnosis and treatment, but serious complication including cataracts , glaucoma , band keratopathy , macular edema and permanent vision loss may result if left untreated. ... In Asian countries the proportion is between 28% and 50%. [31] Uveitis is estimated to be responsible for approximately 10%-20% of the blindness in the United States. [32] See also [ edit ] List of systemic diseases with ocular manifestations Intermediate uveitis References [ edit ] ^ Abdullah Al-Fawaz; Ralph D Levinson (25 Feb 2010).
Overview Uveitis is a form of eye inflammation. It affects the middle layer of tissue in the eye wall (uvea). Uveitis (u-vee-I-tis) warning signs often come on suddenly and get worse quickly. They include eye redness, pain and blurred vision. The condition can affect one or both eyes, and it can affect people of all ages, even children. Possible causes of uveitis are infection, injury, or an autoimmune or inflammatory disease. Many times a cause can't be identified. Uveitis can be serious, leading to permanent vision loss.
A period of experimentation with different treatment regimens is likely to be necessary in order to discover the most appropriate treatment regimen for a given patient. [ citation needed ] Epidemiology [ edit ] In 1982 Lewis et al. reported a group of patients with a chronic asymmetrical sensorimotor neuropathy mostly affecting the arms with multifocal involvement of peripheral nerves. [26] Also in 1982 Dyck et al reported a response to prednisolone to a condition they referred to as chronic inflammatory demyelinating polyradiculoneuropathy. [27] Parry and Clarke in 1988 described a neuropathy which was later found to be associated with IgM autoantibodies directed against GM1 gangliosides. [28] [29] This latter condition was later termed multifocal motor neuropathy [30] This distinction is important because multifocal motor neuropathy responds to intravenous globulin alone while chronic inflammatory demyelinating polyneuropathy responds to intravenous globulin, steroids and plasma exchanges. [31] It has been suggested that multifocal motor neuropathy is distinct from chronic inflammatory demyelinating polyneuropathy and that Lewis-Sumner syndrome is a distinct variant type of chronic inflammatory demyelinating polyneuropathy. [32] The Lewis-Sumner form of this condition is considered a rare disease with only 50 cases reported up to 2004. [33] A total of 90 cases had been reported by 2009. [34] Vaccine Injury Compensation for CIDP [ edit ] The National Vaccine Injury Compensation Program has awarded money damages to patients who came down with CIDP after receiving one of the childhood vaccines listed on the Federal Government's vaccine injury table . ... PMID 12090400 . ^ Hadden, Robert D. M.; Marreno, Fabrizio (2016-12-28). "Switch from intravenous to subcutaneous immunoglobulin in CIDP and MMN: improved tolerability and patient satisfaction" .
Chronic inflammatory demyelinating polyneuropathy (CIDP) is a neurological disorder that causes progressive weakness and impaired sensory function in the legs and arms. Symptoms often include tingling or numbness (first in the toes and fingers), weakness of the arms and legs, loss of deep tendon reflexes, fatigue, and abnormal sensations. Other symptoms may include pain, difficulty swallowing (dysphagia), and double vision ( diplopia ). CIDP is thought to be caused by the immune system mistakenly attacking and damaging the myelin sheath (protective cover of nerve fibers) of the peripheral nerves. CIDP is closely related to Guillain-Barre syndrome (GBS) and is considered the "chronic counterpart" of GBS.
A number sign (#) is used with this entry because a mutation in the PMP22 gene (601097) on chromosome 17 was identified in a single family with the acute (AIDP) and chronic (CIDP) forms of inflammatory demyelinating polyneuropathy. Description Guillain-Barre syndrome (GBS) is an acute inflammatory demyelinating polyneuropathy characterized most commonly by symmetric limb weakness and loss of tendon reflexes. It is a putative autoimmune disorder presenting after an infectious illness, most commonly Campylobacter jejuni, a gram-negative bacterium that causes acute enteritis (Yuki and Tsujino, 1995; Koga et al., 2005). Approximately 1 in 1,000 individuals develops GBS after C. jejuni infection (Nachamkin, 2001). Although rare familial cases have been reported, GBS is considered to be a complex multifactorial disorder with both genetic and environmental factors rather than a disorder following simple mendelian inheritance (Geleijns et al., 2004).
Guillain-Barré syndrome is an autoimmune disorder that affects the nerves. Autoimmune disorders occur when the immune system malfunctions and attacks the body's own tissues and organs. In Guillain-Barré syndrome, the immune response damages peripheral nerves, which are the nerves that connect the central nervous system (the brain and spinal cord) to the limbs and organs. Specifically, the immune response affects a particular part of peripheral nerves called axons, which are the extensions of nerve cells (neurons) that transmit nerve impulses. Guillain-Barré syndrome can affect the neurons that control muscle movement (motor neurons ); the neurons that transmit sensory signals such as pain, temperature, and touch (sensory neurons); or both.
A chronic monophasic, progressive or relapsing symmetric sensorimotor disorder characterized by progressive muscular weakness with impaired sensation, absent or diminished tendon reflexes and elevated cerebrospinal fluid (CSF) proteins. Epidemiology Prevalence is about 1/200,000 children and 1-7/100,000 adults, but it is generally accepted that the frequency is underestimated. Clinical description Onset may occur at any age but is more common in the 5th and 6th decades. Main clinical manifestations include progressive symmetrical weakness in both proximal and distal muscles of lower and/or upper limbs with partial or complete recovery between recurrences, associated with impaired sensation and absent/diminished tendon reflexes. Disease course is relapsing in 30% of cases, chronic and progressive in 60%, and monophasic with full generally permanent recovery in 10%.
A rare inflammatory neuropathy belonging to the clinical spectrum of Guillain-Barré syndrome (GBS). Epidemiology Overall annual incidence of GBS is estimated at between 1/91,000 and 1/55,000. AIDP accounts for around 90% of GBS cases in Europe and North America and thus the term GBS is often synonymous with AIDP in Western countries. The disease occurs in patients of all ages and men are affected about 1.5 times more often than women. Clinical description The clinical course of AIDP is divided into three phases.