- Mitochondrial Complex Ii Deficiency Omim
- Episodic Kinesigenic Dyskinesia 1 Omim
- Aromatase Deficiency Omim
-
Alveolar Capillary Dysplasia With Misalignment Of Pulmonary Veins
Omim
Of 22 infants for whom autopsy reports were available, 18 (82%) had at least 1 additional major structural abnormality, affecting 1 or more of the cardiovascular (5 infants), gastrointestinal (4), genitourinary (4), or musculoskeletal (1) systems; and in 5 (28%) of the 18 infants with additional anomalies, disruption of the normal right-left asymmetry of intrathoracic or intraabdominal organs were noted.
-
Neuropathy, Hereditary Motor And Sensory, Okinawa Type
Omim
., CMT2A1, 118210) in which common CMT2-implicated genes had been excluded. The age at onset ranged from 28 to 40 years, and all patients presented with slowly progressive symmetrical muscle atrophy and weakness predominantly affecting the distal parts of the limbs and causing gait difficulties in most patients.

-
Dental Anomalies And Short Stature
Omim
Mapping In a consanguineous Pakistani family segregating autosomal recessive selective tooth agenesis and short stature, in which mutation in the MSX1 (142983) and PAX9 (167416) genes had been excluded, Noor et al. (2009) performed genomewide linkage analysis and microsatellite marker genotyping. The authors identified a 28-Mb autozygous region spanning the centromere of chromosome 11 that was shared among the 4 affected sibs but not among unaffected members of the family.
-
Aicardi Syndrome
Omim
In 18 girls with Aicardi syndrome identified through a survey of neurologists, geneticists, and ophthalmologists, Donnenfeld et al. (1989) found complete agenesis of the corpus callosum in 72% and partial agenesis in 28%. Costovertebral defects including hemivertebrae, scoliosis, and absent or malformed ribs were present in 39%.

-
Robinow Syndrome, Autosomal Dominant 1
Omim
INHERITANCE - Autosomal dominant GROWTH Height - Short stature (postnatal onset) (81%) HEAD & NECK Head - Macrocephaly (64%) - Large anterior fontanel Face - Frontal bossing (79%) - Long philtrum (65%) - Micrognathia (57%) - Midface hypoplasia (81%) - Retrognathia (44%) - Flat facial profile Ears - Posteriorly rotated ears - Low-set ears (28%) Eyes - Hypertelorism (100%) - Wide palpebral fissures (50%) - Upslanting palpebral fissures (37%) - Epicanthal folds (39%) - Prominent eyes (37%) - Long eyelashes (54%) Nose - Short, upturned nose (83%) - Depressed nasal bridge (78%) - Broad nasal bridge (97%) - Anteverted nares (96%) Mouth - Triangular mouth (65%) - Downturned corners of mouth (63%) - Thin upper lip (50%) - Gingival hyperplasia (36%) - Macroglossia - Bifid tongue (39%) - Narrow palate (46%) - High-arched palate (52%) - Short palate - Flat palate - Cleft lip/palate (35%) - Abnormal uvula (89%) Teeth - Crowded teeth (49%) - Infranumerary teeth (16%) - Delayed dental eruption - Wide retromolar ridge Neck - Short neck (29%) CARDIOVASCULAR Heart - Right ventricular outlet obstruction CHEST Ribs Sternum Clavicles & Scapulae - Pectus excavatum (44%) ABDOMEN External Features - Umbilical hernia (32%) - Abnormal umbilicus GENITOURINARY External Genitalia (Male) - Small penis (84%) - Inguinal hernia (17%) External Genitalia (Female) - Small clitoris (46%) - Small labia minora (50%) - Small labia majora (35%) Internal Genitalia (Male) - Cryptorchidism (72%) Kidneys - Renal anomalies (27%) - Renal duplication - Hydronephrosis SKELETAL - Delayed bone age Limbs - Mesomelic limb shortening (80%) - Rhizomelic limb shortening (35%) Hands - Small hands (62%) - Brachydactyly (81%) - Clinodactyly (70%) - Broad thumbs (36%) - Brachymesophalangism V - Bifid terminal phalanges Feet - Broad toes (33%) - Bifid terminal phalanges SKIN, NAILS, & HAIR Skin - Nevus flammeus Nails - Nail dysplasia (22%) NEUROLOGIC Central Nervous System - Developmental delay (20%) - Mental retardation (20%) MISCELLANEOUS - See also autosomal recessive Robinow syndrome ( 268310 ) MOLECULAR BASIS - Caused by mutation in the wingless-type MMTV integration site family, member 5A gene (WNT5A, 164975.0001 ) ▲ CloseWNT5A, DVL3, DVL1, FZD2, ATR, RAF1, CEP63, CENPJ, LZTR1, SOS1, RIT1, PTPN11, KRAS, DNAAF4, ROR2, PTCH1, SYK, NTRK1, IGF1, GH1
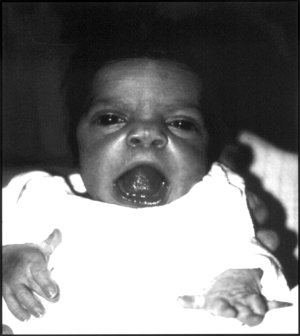
- Pyruvate Kinase Deficiency Of Red Cells Omim
-
Argininosuccinic Aciduria
Omim
McInnes et al. (1984) performed complementation analysis in a search for genetic heterogeneity in this disorder. In 20 of 28 fibroblast strains cultured from patients with ASL deficiency, partial complementation was observed, with 2- to 10-fold increases in lyase activity.

- Spondyloepiphyseal Dysplasia Congenita Omim
-
Thiourea Tasting
Omim
Population Genetics Kim and Drayna (2004) stated that the frequency of PTC nontasters in Caucasians is approximately 28%. Henkin and Gillis (1977) observed that 2 of 8 people served a pie made from berries of the tree Antidesma bunius, a popular fruit in southeast Asia and Florida, found their dessert bitter and inedible, whereas the remainder found it edible and sweet.
-
Idiopathic Orbital Inflammatory Disease
Wikipedia
Berlin: Julius Springer. p. 251–253, 1930 ^ a b c LeBedis CA, Sakai O: Nontraumatic Orbital Conditions: Diagnosis with CT and MR Imaging in the Emergent Setting. RadioGraphics. 28(6):1741–1753, 2008 ^ a b c d Rubin PAD, Foster CS: Etiology and Management of Idiopathic Orbital Inflammation.

-
Chromosome 17q12 Deletion Syndrome
Omim
Renal Cysts and Diabetes Syndrome Bellanne-Chantelot et al. (2005) studied 40 unrelated patients with a clinical phenotype consistent with maturity-onset diabetes of the young type 5 (MODY5; 137920), in which patients present with renal cysts and diabetes, and found molecular alterations of the TCF2 gene (HNF1B; 189907) in 28 (70%). Point mutations were identified in 18 patients and 10 had gross genomic rearrangements, which in 9 patients involved a deletion of at least 1.2 Mb, delimited by the TCF2 and TRIP3 (604500) genes and encompassing 7 other genes and 2 predicted proteins.
-
X-Linked Hypophosphatemia
Wikipedia
Journal of Bone and Mineral Research . 28 (3): 688–699. doi : 10.1002/jbmr.1766 .PHEX, CLCN5, FGF23, L1CAM, PTH, DMP1, GH1, BGLAP, ENPP1, RAPGEF5, SPP1, KL, MEPE, SOST, TBX1, TYRP1, SOX9, ACTB, TBPL1, SLC17A1, DKK1, FAM20C, PRRT2, PTRH1, SLC34A3, SKA1, SFTPA1, SLC34A1, PRKCB, SCG5, ETFA, AVPR2, BPI, CALCA, CALCR, COX8A, CTNNB1, CTSK, FGF2, SFRP1, FRZB, ANOS1, MMP13, MUC1, NT5E, PRKCA, AR, POTEF

-
Hypothyroidism, Central, With Testicular Enlargement
Omim
Transient neonatal hypocortisolism was present in 6 (21%) of 28 male newborns for whom data was available, resolving within a few years in all.
-
Chemotherapy-Induced Peripheral Neuropathy
Wikipedia
Vergo; Axel Grothey; Julia Minocha; Jonathan Cotliar (28 October 2013). "Management of systemic treatment-induced toxicities" .MMP9, HDAC6, HMGB1, TLR4, HSPB1, HSPB2, ARHGEF10, TNF, SARM1, HSPB3, NGF, BDNF, SRI, CX3CL1, TNS1, TP53, TYROBP, SRL, RPS6KB2, RAG2, VEGFA, AKT1, TRPV1, TLR9, HCN1, EPHA6, SPNS1, PRX, CEP72, TREM2, NCS1, NR1I2, RAPGEF3, SIGMAR1, ABCC4, NAMPT, TRPA1, PIK3CG, PTGS2, PIK3CA, PIK3CD, PIK3CB, BPI, CALCA, CASP3, TNFRSF8, CHRNA4, MAPK14, CX3CR1, DDIT3, SARDH, EPHA5, EPHA8, ERBB2, F3, FAAH, PTK2B, GPX7, GSK3B, HCRT, HIF1A, HSPA4, IL1B, IL6, IL17A, MAPT, NARS1, NEFL, OPRM1, P2RX3, ANK1, MIR338
-
Muscle Imbalance
Wikipedia
.; Friedman, Neil J.; II, Roberto Pineda II (2014-02-28). The Massachusetts Eye and Ear Infirmary Illustrated Manual of Ophthalmology .

-
Invasive Candidiasis
Wikipedia
Emergent species [ edit ] Candida auris is an emerging multidrug-resistant yeast that can cause invasive candidiasis and is associated with high mortality. [6] It was first described in 2009. [6] Since then, C. auris infections, specifically fungemia, have been reported from South Korea, India, South Africa, Kuwait, Colombia, Venezuela, Pakistan, the United Kingdom and the United States. [6] The strains isolated in each region are genetically distinct, indicating that this species is emerging in different locations. [6] The reason for this pattern is unknown. [6] Risk factors [ edit ] Patients with the following conditions, treatments or situations are at increased risk for invasive candidiasis. [4] [2] [7] Critical illness Long-term intensive care unit stay Abdominal surgery (aggravated by anastomotic leakage or repeat laparotomies ) Immunosuppressive diseases Acute necrotizing pancreatitis Malignant hematologic disease Solid-organ transplantation Hematopoietic stem cell transplantation Solid- organ tumors Neonates (especially low birth weight and preterm infants) Broad-spectrum antibiotic treatment Central venous catheter Internal prosthetic device Total parenteral nutrition Hemodialysis Glucocorticoid use Chemotherapy Noninvasive Candida colonization (particularly if multifocal) Transmission [ edit ] Invasive candidiasis is a nosocomial infection with the majority of cases associated with hospital stays. [4] Diagnosis [ edit ] Because many Candida species are part of the human microbiota , their presence in the mouth, the vagina, sputum, urine, stool, or skin is not definitive evidence for invasive candidiasis. [2] Positive culture of Candida species from normally sterile sites, such as blood, cerebrospinal fluid , pericardium , pericardial fluid , or biopsied tissue , is definitive evidence of invasive candidiasis. [2] Diagnosis by culturing allows subsequent susceptibility testing of causative species. [8] [7] Sensitivity of blood culture is far from ideal, with a sensitivity reported to be between 21 and 71%. [7] Additionally, whereas blood culture can establish a diagnosis during fungemia , the blood may test negative for deep-seated infections because candida may have been successfully cleared from the blood. [7] Diagnosis of invasive candidiasis is supported by histopathologic evidence (for example, yeast cells or hyphae ) observed in specimens of affected tissues. [2] Additionally, elevated serum β-glucan can demonstrate invasive candidiasis while a negative test suggests a low likelihood of systemic infection. [9] [2] The emergence of multidrug-resistant C. auris as a cause of invasive candidiasis has necessitated additional testing in some settings. [6] C. auris -caused invasive candidiasis is associated with high mortality. [6] Many C. auris isolates have been found to be resistant to one or more of the three major antifungal classes (azoles, echinocandins, and polyenes) with some resistant to all three classes – severely limiting treatment options. [6] Biochemical-based tests currently used in many laboratories to identify fungi, including API 20C AUX and VITEK-2 , cannot differentiate C. auris from related species (for example, C. auris can be identified as C. haemulonii ). [6] Therefore, the Centers for Disease Control and Prevention recommends using a diagnostic method based on matrix-assisted laser desorption/ionization-time of flight mass spectrometry or a molecular method based on sequencing the D1-D2 region of the 28s rDNA to identify C. auris in settings where it may be present. [6] Prevention [ edit ] Preventive antifungal treatment is supported by studies, but only for specific high-risk groups in intensive care units with conditions that put them at high risk for the disease. [7] For example, one group would be patients recovering from abdominal surgery that may have gastrointestinal perforations or anastomotic leakage . [7] Antifungal prophylaxis can reduce the incidence of fungemia by approximately 50%, but has not been shown to improve survival. [7] A major challenge limiting the number of patients receiving prophylaxis to only those that can potentially benefit, thereby avoiding the creation of selective pressure that can lead to the emergence of resistance . [7] Treatment [ edit ] Antifungals are used for treatment with the specific type and dose depending on the patient's age, immune status, and specifics of the infection.
-
Episcleritis
Wikipedia
Most cases of episcleritis resolve within 7–10 days. [1] The nodular type is more aggressive and takes longer to resolve. [1] Although rare, some cases may progress to scleritis. [12] However, in general, episcleritis does not cause complications in the eye. [12] Smoking tobacco delays the response to treatment in patients with episcleritis. [13] Epidemiology [ edit ] While episcleritis is a common disease, [14] its exact prevalence and incidence are unknown. [5] It typically affects young [14] or middle aged women. [5] The diffuse form of episcleritis (70%) is more common than the nodular form (30%). [5] One retrospective study found 28 percent of individuals with episcleritis experienced recurrent episodes of the disease. [15] References [ edit ] ^ a b c d e Heath, Greg (10 February 2010).











