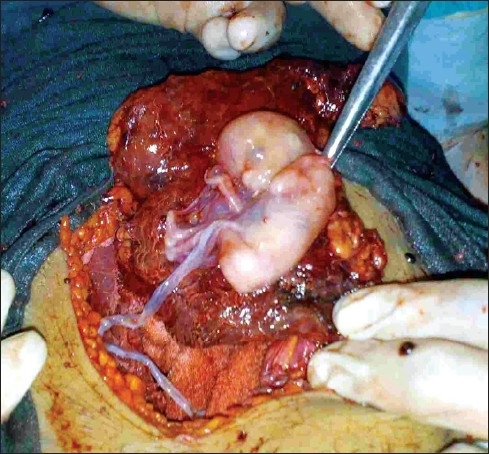
Advanced abdominal pregnancy [ edit ] Advanced abdominal pregnancy refers to situations where the pregnancy continues past 20 weeks of gestation (versus early abdominal pregnancy < 20 weeks). [2] [27] In those situations, live births have been reported in the lay press where the babies are not uncommonly referred to as 'Miracle babies'. [28] [29] A patient may carry a dead fetus but will not go into labor.
Psychology researchers surveyed 253 undergraduate students at the University of Albany and found that not only is social media (particularly Facebook) itself potentially addictive, those who use it may also be at greater risk for substance abuse. [24] Biomolecular mechanisms [ edit ] Main article: ΔFosB ΔFosB , a gene transcription factor, has been identified as playing a critical role in the development of addictive states in both behavioral addictions and drug addictions. [4] [5] [8] Overexpression of ΔFosB in the nucleus accumbens is necessary and sufficient for many of the neural adaptations seen in drug addiction; [4] it has been implicated in addictions to alcohol , cannabinoids , cocaine , nicotine , phenylcyclidine , and substituted amphetamines [4] [25] [26] [27] as well as addictions to natural rewards such as sex, exercise, and food. [5] [8] A recent study also demonstrated a cross-sensitization between drug reward (amphetamine) and a natural reward (sex) that was mediated by ΔFosB. [28] Besides increased ΔFosB expression in the nucleus accumbens, there are many other correlations in the neurobiology of behavioral addictions with drug addictions.
McGivney Recurrences [ edit ] 23 January 1891 Prince Baudouin of Belgium [a] 10 February 1891 Sofya Kovalevskaya 18 March 1891 William Herndon 8 May 1891 Helena Blavatsky 15 May 1891 Edwin Long 3 June 1891 Oliver St John 9 June 1891 Henry Gawen Sutton 1 July 1891 Frederic Edward Manby 20 December 1891 Grisell Baillie 28 December 1891 William Arthur White 8 January 1892 John Tay 10 January 1892 John George Knight 12 January 1892 Jean Louis Armand de Quatrefages de Bréau 14 January 1892 Prince Albert Victor, Duke of Clarence and Avondale , grandson of Queen Victoria 17 January 1892 Charles A.
According to Joseph Patrick Byrne, "By war's end, typhus may have killed more than 10 percent of the total German population, and disease in general accounted for 90 percent of Europe's casualties." [27] 19th century [ edit ] During Napoleon 's retreat from Moscow in 1812, more French soldiers died of typhus than were killed by the Russians. [28] A major epidemic occurred in Ireland between 1816 and 1819, during the famine caused by a worldwide reduction in temperature known as the Year Without a Summer .
The fatigue is not due to exercise and is not relieved by rest. [28] Through numerous studies, it has been shown that people with chronic fatigue syndrome have an integral central fatigue component. [1] In one study, the subjects' skeletal muscles were checked to ensure they had no defects that prevented their total use.
HPIVs is believed to be associated with 10% of all LRI cases, thus remaining a significant cause of mortality. [12] Risk factors [ edit ] Numerous factors have been suggested and linked to a higher risk of acquiring the infection, inclusive of malnutrition , vitamin A deficiency , absence of breastfeeding during the early stages of life, environmental pollution and overcrowding. [27] Prevention [ edit ] Despite decades of research, no vaccines currently exist. [28] Recombinant technology has however been used to target the formation of vaccines for HPIV-1, -2 and -3 and has taken the form of several live-attenuated intranasal vaccines.
Neutropenic fever in individuals treated for cancer has a mortality of 4–30%. [28] Epidemiology [ edit ] Neutropenia is usually detected shortly after birth, affecting 6% to 8% of all newborns in neonatal intensive care units (NICUs).
The UK National Health Service advises 150 minutes (2 hours and 30 minutes) of moderate-intensity aerobic activity per week to help prevent hypertension. [21] Pathophysiology [ edit ] Main article: Pathophysiology of hypertension A diagram explaining factors affecting arterial pressure Cardiac output and peripheral resistance are the two determinants of arterial pressure and so blood pressure is normally dependent on the balance between cardiac output and peripheral resistance. [28] Cardiac output is determined by stroke volume and heart rate ; stroke volume is related to myocardial contractility and to the size of the vascular compartment .
A number sign (#) is used with this entry because variations in many genes contribute to essential hypertension. For information on genetic heterogeneity of essential hypertension, see the MAPPING section. Description The Pickering school held that blood pressure has a continuous distribution, that multiple genes and multiple environmental factors determine the level of one's blood pressure just as the determination of stature and intelligence is multifactorial, and that 'essential hypertension' is merely the upper end of the distribution (Pickering, 1978). In this view the person with essential hypertension is one who happens to inherit an aggregate of genes determining hypertension (and also is exposed to exogenous factors that favor hypertension). The Platt school took the view that essential hypertension is a simple mendelian dominant trait (Platt, 1963).
Hypertension is abnormally high blood pressure in the arteries, which are the blood vessels that carry blood from the heart to the rest of the body. As the heart beats, it forces blood through the arteries to deliver nutrients and oxygen to the rest of the body. The strength of the blood pushing against the artery walls is blood pressure, which is measured in units called millimeters of mercury (mmHg). The top number in a blood pressure reading is the pressure when the heart pumps (systolic blood pressure), and the bottom number is the pressure between heart beats (diastolic blood pressure). In adults, a normal blood pressure measurement is about 120/80 mmHg. Blood pressure is considered high when the measurement is 130/80 mmHg or greater.
The approximate location of the course of the duct is the middle third of this line. [25] Nerves and muscles may be trapped by broken bones; in these cases the bones need to be put back into their proper places quickly. [4] For example, fractures of the orbital floor or medial orbital wall of the eye can entrap the medial rectus or inferior rectus muscles . [26] In facial wounds, tear ducts and nerves of the face may be damaged. [3] Fractures of the frontal bone can interfere with the drainage of the frontal sinus and can cause sinusitis . [27] Infection is another potential complication, for example when debris is ground into an abrasion and remains there. [4] Injuries resulting from bites carry a high infection risk. [3] Epidemiology [ edit ] As many as 50–70% of people who survive traffic accidents have facial trauma. [3] In most developed countries, violence from other people has replaced vehicle collisions as the main cause of maxillofacial trauma; however in many developing countries traffic accidents remain the major cause. [8] Increased use of seat belts and airbags has been credited with a reduction in the incidence of maxillofacial trauma, but fractures of the mandible (the jawbone) are not decreased by these protective measures. [9] The risk of maxillofacial trauma is decreased by a factor of two with use of motorcycle helmets. [9] A decline in facial bone fractures due to vehicle accidents is thought to be due to seat belt and drunk driving laws, strictly enforced speed limits and use of airbags. [7] In vehicle accidents, drivers and front seat passengers are at highest risk for facial trauma. [9] Facial fractures are distributed in a fairly normal curve by age, with a peak incidence occurring between ages 20 and 40, and children under 12 suffering only 5–10% of all facial fractures. [28] Most facial trauma in children involves lacerations and soft tissue injuries. [4] There are several reasons for the lower incidence of facial fractures in children: the face is smaller in relation to the rest of the head, children are less often in some situations associated with facial fractures such as occupational and motor vehicle hazards , there is a lower proportion of cortical bone to cancellous bone in children's faces, poorly developed sinuses make the bones stronger, and fat pads provide protection for the facial bones. [4] Head and brain injuries are commonly associated with facial trauma, particularly that of the upper face; brain injury occurs in 15–48% of people with maxillofacial trauma. [29] Coexisting injuries can affect treatment of facial trauma; for example they may be emergent and need to be treated before facial injuries. [11] People with trauma above the level of the collar bones are considered to be at high risk for cervical spine injuries (spinal injuries in the neck) and special precautions must be taken to avoid movement of the spine, which could worsen a spinal injury . [23] References [ edit ] ^ a b c d e f g h i j Seyfer AE, Hansen JE (2003) . pp. 423–24. ^ a b c d e Munter DW, McGurk TD (2002).
For example, the disease can have devastating effects on the careers, caretakers and family members of patients. [27] [28] Those who are working lose their ability to perform their jobs competently, and are forced into early retirement.
Routine screening for CTEPH after PE is not recommended because a significant number of CTEPH cases develops in the absence of previous acute symptomatic PE. [6] In addition, approximately 25% of patients with CTEPH do not present with a clinical history of acute PE. [1] The median age of patients at diagnosis is 63 years (there is a wide age range, but paediatric cases are rare), and both genders are equally affected. [1] [28] [29] References [ edit ] ^ a b c d e f Pepke-Zaba, Joanna; Delcroix, Marion; Lang, Irene; Mayer, Eckhard; Jansa, Pavel; Ambroz, David; Treacy, Carmen; D'Armini, Andrea M.; Morsolini, Marco (2011-11-01).
Chronic thromboembolic pulmonary hypertension (CTEPH) is a unique pulmonary vascular disease caused by chronic block of the major lung arteries . Signs and symptoms commonly include progressive breathing difficulties (dyspnea) on exertion, fatigue, palpitations, loss of consciousness ( syncope), or swelling (edema). The disease may appear a few months or many years after the sudden blockage in a lung artery by a blood clot (acute pulmonary embolism ). However, up to 60% of patients have no history of acute pulmonary embolism. Some people with this disease may have clotting problems . Research suggests there may be a genetic predisposition leading to abnormal vascular healing after pulmonary embolism in susceptible individuals, but no specific gene mutations have been identified in CTEPH.
Chronic thromboembolic pulmonary hypertension (CTEPH) is characterized by the persistence of thromboemboli in the form of organized tissue obstructing the pulmonary arteries. The consequence is an increase in pulmonary vascular resistance (PVR) resulting in pulmonary hypertension (PH) and progressive right heart failure. Epidemiology The true prevalence is unknown: CTEPH is a rare disease but recent reports suggest that it is underdiagnosed. Clinical description Patients commonly present with progressive dyspnea on exertion with or without signs of right heart dysfunction including fatigue, palpitations, syncope, or edema. A period between the initial event (acute embolism) and the development of clinical signs is common and may last from a few months to many years.
Clinical Features Chronic thromboembolic pulmonary hypertension (CTEPH) is a rare disease characterized by persistent pulmonary embolism resulting in pulmonary hypertension and consequent right heart failure. The estimated annual incidence of CTEPH is 500 to 2,500 patients in the U.S. and the estimated prevalence in Japan is about 450 patients. Although deep vein thrombosis (DVT) may be a predisposing factor, many patients do not have concurrent DVT, suggesting that there are different clinical categories, namely DVT-positive CTEPH and DVT-negative CTEPH (Kominami et al., 2009). Mapping Among 160 Japanese patients with CTEPH, 99 without DVT and 61 with DVT, and 380 controls, Kominami et al. (2009) identified multiple associations between DVT-negative CTEPH and markers on chromosome 6p21.3. The associations were for marker DPB1*0202 of the HLA-DPB1 gene (142858) (OR, 5.07; corrected p value = 0.00014), IKBLp*03 of the NFKBIL1 gene (601022) (OR, 2.33; corrected p value = 0.033), and HLA-B*5201 of the HLA-B gene (142830) (OR, 2.47; corrected p value = 0.016).
Marker chromosomes were more often confirmed in the fetus than trisomies. [3] [10] For example, of 28 cases of mosaic polyploidy detected on CVS, fetal mosaicism was confirmed in only one case.
Current chemotherapy consists of continuous infusion 5-FU over four days with bolus mitomycin given concurrently with radiation. 5-FU and cisplatin are recommended for metastatic anal cancer. [28] Metastatic or recurrent disease [ edit ] 10 to 20% of patients treated for anal cancer will develop distant metastatic disease following treatment. [29] Metastatic or recurrent anal cancer is difficult to treat, and usually requires chemotherapy .
Anal cancer is a rare form of cancer that occurs due to abnormal and uncontrolled cell growth in the anus . Signs and symptoms of the condition include rectal bleeding ; a lump in or near the anus; anal pain; itching; changes in bowel habits; and/or swollen lymph nodes . In most cases, the underlying cause of anal cancer is unknown. There appears to be a link between anal cancer and the human papillomavirus (HPV) infection. Other risk factors for anal cancer include HIV infection, sexual activity, smoking, and a weakened immune system. The best treatment options for anal cancer depend on many factors including the stage of the condition, the location of the tumor and if there are associated conditions (i.e.
Overview Anal cancer is an uncommon type of cancer that occurs in the anal canal. The anal canal is a short tube at the end of your rectum through which stool leaves your body. Anal cancer can cause signs and symptoms such as rectal bleeding and anal pain. Most people with anal cancer are treated with a combination of chemotherapy and radiation. Though combining anal cancer treatments increases the chance of a cure, the combined treatments also increase the risk of side effects.
"A new protein substitute for adolescents and adults with maple syrup urine disease (MSUD)". J. Inherit. Metab. Dis . 28 (5): 665–672. doi : 10.1007/s10545-005-0061-6 .
Mean and 25th to 75th Percentile Range Nutrient Intakes (per kg-day) by Age Group View in own window Nutrient Age in Months (# of Persons) 0-2 (31) 3-5 (18) 6-8 (21) 9-12 (18) 13-18 (21) 19-24 (18) 25-36 (32) Mean Nutrient Intake per kg-day (25th to 75th %ile range) Leucine (mg) 72 (64-84) 58 (47-68) 44 (37-51) 35 (30-41) 33 (26-39) 27 (22-28) 21 (20-25) Energy (kcal) 111 (103-119) 99 (94-107) 89 (82-99) 78 (71-87) 67 (55-77) 57 (49-71) 38 (39-51) Total protein (g) 2.4 (2.1-2.6) 2.3 (1.9-2.6) 2.2 (1.8-2.5) 2.0 (1.5-2.5) 2.1 (1.8-2.4) 2 (1.7-2.3) 1.5 (1.5-2.9) Leucine/energy ratio (mg/kcal) 0.65 (0.57-0.72) 0.58 (0.48-0.66) 0.50 (0.43-0.56) 0.46 (0.37-0.53) 0.50 (0.43-0.58) 0.48 (0.38-0.58) 0.54 (0.44-0.55) Strauss et al [2010]; with permission from Elsevier Data derived from Mennonite children from birth to age 4 years with classic MSUD Mild illness.
A number sign (#) is used with this entry because of evidence that a mild variant of maple syrup urine disease (MSUDMV) is caused by homozygous mutation in the PPM1K gene (611065) on chromosome 4q22. One such family has been reported. Description The mild variant of MSUD is characterized by increased plasma levels of branched-chain amino acids (BCAA) apparent at birth. Treatment with a low-protein diet free of BCAA can result in normal psychomotor development and lack of metabolic episodes; however, plasma levels of BCAA may remain elevated (summary by Oyarzabal et al., 2013). For a general description and a discussion of genetic heterogeneity of maple syrup urine disease, see 248600. Clinical Features Oyarzabal et al. (2013) reported a 21-year-old woman with a mild variant of maple syrup urine disease.
A rare inherited disorder of branched-chain amino acid metabolism classically characterized by poor feeding, lethargy, vomiting and a maple syrup odor in the cerumen (and later in urine) noted soon after birth, followed by progressive encephalopathy and central respiratory failure if untreated. The four overlapping phenotypic subtypes are: classic, intermediate, intermittent and thiamine-responsive MSUD. Epidemiology The estimated prevalence is around 1/150,000 live births, from published and unpublished newborn screening data. Clinical description Classic MSUD presents in the first days of life with poor feeding and drowsiness followed by a worsening encephalopathy with lethargy, intermittent apnea, stereotypic movements ("fencing" and ''bicycling") and opisthotonus. Coma and central respiratory failure supervene 7 to 10 days after birth.
Maple syrup urine disease (MSUD) occurs when the body is unable to breakdown certain parts of proteins. This leads to the build-up of toxic substances that can cause organ and brain damage. There are several forms of MSUD. The most common is the classic or infantile form. Symptoms of the classic form of MSUD start in early infancy and include poor feeding, irritability, extra sleepiness, and muscle spasms. If untreated, respiratory failure (lack of oxygen getting to the blood) may occur.
Reverse isolation involves the use of laminar air flow and mechanical barriers (to avoid physical contact with others) to isolate the patient from any harmful pathogens present in the external environment. [23] A non-curative treatment for patients with ADA-SCID is enzyme replacement therapy, in which the patient is injected with polyethyleneglycol-coupled adenosine deaminase (PEG-ADA) which metabolizes the toxic substrates of the ADA enzyme and prevents their accumulation. [20] Treatment with PEG-ADA may be used to restore T cell function in the short term, enough to clear any existing infections before proceeding with curative treatment such as a bone marrow transplant. [24] Epidemiology [ edit ] The most commonly quoted figure for the prevalence of SCID is around 1 in 100,000 births, although this is regarded by some to be an underestimate of the true prevalence; [25] some estimates predict that the prevalence rate is as high as 1 in 50,000 live births. [3] A figure of about 1 in 65,000 live births has been reported for Australia . [26] Due to the genetic nature of SCID, a higher prevalence is found in areas and cultures among which there is a higher rate of consanguineous mating. [27] A study conducted upon Moroccan SCID patients reported that inbreeding parenting was observed in 75% of the families. [28] Recent studies indicate that one in every 2,500 children in the Navajo population inherit severe combined immunodeficiency.
Severe combined immunodeficiency (SCID) comprises a group of rare monogenic primary immunodeficiency disorders characterized by a lack of functional peripheral T lymphocytes resulting in early-onset severe respiratory infections and failure to thrive. They are classified according to immunological phenotype into SCID with absence of T cells but presence of B cells (T-B+ SCID) or SCID with absence of both (T-B- SCID) (see these terms). Both of these groups include several forms, with or without natural killer (NK) cells. Epidemiology Overall incidence is estimated at about 1/50,000 live births, with regional differences and higher incidences among populations with a higher consanguinity rate. The disease affects more males because of the X-linked variant (SCID T-B+ due to gamma chain deficiency; see this term) that represents about 30% of SCID cases in Western countries.
Severe combined immunodeficiencies (SCID) are inherited immune system disorders characterized by abnormalities with responses of both T cells and B cells (specific types of white blood cells needed for immune system function). Common signs and symptoms include an increased susceptibility to infections including ear infections; pneumonia or bronchitis; oral thrush; and diarrhea. Due to recurrent infections, children with SCID do not grow and gain weight as expected ( failure to thrive ). SCID may be caused by mutations in any of several genes and can be inherited in an X-linked recessive (most commonly) or autosomal recessive manner. The most common type of SCID is called X-linked severe combined immunodeficiency (XSCID).
Both are available in Europe and in many other parts of the world, but treatment costs remain very high. [27] While increasing evidence shows that long-term enzyme therapy can halt the disease progression, the importance of adjunctive therapies should be emphasized and the possibility of developing an oral therapy pushes research forward into active site specific chaperones. [14] Besides these drugs, a gene therapy treatment is in clinical trials, [28] [29] with the technology licensed to AvroBio. [30] Other treatments under research include: plant-based ERT from Protalix, substrate reduction therapy from Sanofi-Genzyme, bio-better ERT from Codexis, and a gene editing solution from Sangamo. [31] Organ-specific treatment [ edit ] Pain associated with Fabry disease may be partially alleviated by enzyme replacement therapy in some patients, but pain management regimens may also include analgesics , anticonvulsants , and nonsteroidal anti-inflammatory drugs , though the latter are usually best avoided in kidney disease.
Clinical Management Dauvilliers et al. (2009) reported a 28-year-old woman with narcolepsy who had complete reversal of symptomatology after intravenous immunoglobulin (IVIg) infusion.
For a phenotypic description and a discussion of genetic heterogeneity of narcolepsy, see 161400. Mapping Miyagawa et al. (2008) conducted a genomewide association study in 222 Japanese individuals with narcolepsy and 389 Japanese controls, with replication of top hits in 159 Japanese individuals with narcolepsy and 190 Japanese controls, followed by the testing of 424 Koreans, 785 individuals of European descent, and 184 African Americans. A T-to-C SNP (rs5770917) located between the CPT1B (601987) and CHKB (612395) genes on chromosome 22q13 was significantly associated with narcolepsy in Japanese (odds ratio (OR) = 1.79, p = 4.4 x 10(-7)). A metaanalysis in the 4 populations yielded an OR of 1.63 (p = 5.9 x 10(-8)). Real-time quantitative PCR assays in white blood cells indicated decreased CPT1B and CHKB expression in individuals with the C allele, suggesting that a genetic variant regulating CPT1B or CHKB expression may be associated with narcolepsy.
Narcolepsy is a chronic brain disorder that involves poor control of sleep-wake cycles. People with narcolepsy have episodes of extreme daytime sleepiness and sudden, irresistible bouts of sleep (called "sleep attacks") that can occur at any time, and may last from seconds or minutes. Other signs and symptoms may include cataplexy (a sudden loss of muscle tone that makes a person go limp or unable to move); vivid dream-like images or hallucinations; and/or total paralysis just before falling asleep or after waking-up. Narcolepsy may have several causes, the most common being low levels of the neurotransmitter hypocretin (for various possible reasons). The disorder is usually sporadic but some cases are familial. There is no cure, but some symptoms can be managed with medicines and lifestyle changes.
For a phenotypic description and a discussion of genetic heterogeneity of narcolepsy, see 161400. Mapping In a genomewide linkage search for narcolepsy in 8 Japanese families with 21 DR2-positive patients (14 narcoleptic cases with cataplexy and 7 with an incomplete form of narcolepsy), Nakayama et al. (2000) found a lod score of 3.09 at marker D4S2987 in the 4p13-q21 region.
A rare neurologic disease characterized by excessive daytime sleepiness associated with uncontrollable sleep urges and cataplexy (sudden loss of muscle tone while awake, often triggered by pleasant emotions). Epidemiology Narcolepsy type 1 prevalence is estimated between 1/2,000 and 1/5,000. Clinical description The age of onset varies between 10 and 30 years old and symptoms are lifelong. The average time between the age of appearance of the symptoms and the diagnosis is still very long, 10 years. Other, non specific, clinical signs include hypnagogic hallucinations, sleep paralysis, disturbed nocturnal sleep, and weight gain, especially in children.
A 2009 study found a strong association with polymorphisms in the TRAC gene locus ( dbSNP IDs rs1154155, rs12587781, and rs1263646). [14] A 2013 review article reported additional but weaker links to the loci of the genes TNFSF4 (rs7553711), Cathepsin H (rs34593439), and P2RY11 - DNMT1 (rs2305795). [20] Another gene locus that has been associated with narcolepsy is EIF3G (rs3826784). [21] H1N1 vaccine [ edit ] A link between GlaxoSmithKline 's H1N1 flu vaccine Pandemrix and narcolepsy has been found in both children and adults. [22] Finland's National Institute of Health and Welfare recommended that Pandemrix vaccinations be suspended pending further investigation into narcolepsy. [23] [24] Pathophysiology [ edit ] Loss of neurons [ edit ] Orexin, otherwise known as hypocretin, is a neuropeptide that acts within the brain to regulate appetite and wakefulness as well as a number of other cognitive and physiological processes. [15] [25] [26] Loss of these orexin-producing neurons causes narcolepsy and most individuals with narcolepsy have a reduced number of these neurons in their brains. [15] [16] [19] Selective destruction of the HCRT/OX neurons with preservation of proximate structures suggests a highly specific autoimmune pathophysiology. [27] Cerebrospinal fluid HCRT-1/OX-A is undetectable in up to 95% of patients with type 1 narcolepsy. [27] The system which regulates sleep, arousal, and transitions between these states in humans is composed of three interconnected subsystems: the orexin projections from the lateral hypothalamus , the reticular activating system , and the ventrolateral preoptic nucleus . [16] In narcoleptic individuals, these systems are all associated with impairments due to a greatly reduced number of hypothalamic orexin projection neurons and significantly fewer orexin neuropeptides in cerebrospinal fluid and neural tissue, compared to non-narcoleptic individuals. [16] Those with narcolepsy generally experience the REM stage of sleep within five minutes of falling asleep, while people who do not have narcolepsy (unless they are significantly sleep deprived) [28] do not experience REM until after a period of slow-wave sleep , which lasts for about the first hour or so of a sleep cycle. [1] Disturbed sleep states [ edit ] The neural control of normal sleep states and the relationship to narcolepsy are only partially understood. ... ClinicalTrials.gov. Archived from the original on 28 October 2012. ^ a b Lynn Marie Trotti, MD (15 June 2010). ... "Use of subcutaneous flumazenil preparations for the treatment of idiopathic hypersomnia: A case report". Journal of Psychopharmacology . 28 (7): 703–6. doi : 10.1177/0269881114523865 . ... "Improvement in daytime sleepiness with clarithromycin in patients with GABA-related hypersomnia: Clinical experience". Journal of Psychopharmacology . 28 (7): 697–702. doi : 10.1177/0269881113515062 . ... The Lancet. Neurology . 14 (3): 318–28. doi : 10.1016/S1474-4422(14)70218-2 .
For a phenotypic description and a discussion of genetic heterogeneity of narcolepsy, see 161400. Mapping Hallmayer et al. (2009) performed genomewide association studies on 807 patients with narcolepsy and 1,074 controls of mixed European ancestry. All cases were HLA-DQB1*0602 positive and all had clear-cut cataplexy. Among the 23% with hypocretin-1 (HCRT; 602358) measurements, all were found to be deficient. The authors identified significant association with 3 SNPs in linkage disequilibrium on chromosome 14q11.2: rs1154155 (p = 1.9 x 10(-13)), rs12587781 (p = 3.03 x 10(-13)), and rs1263646 (p = 4.86 x 10(-12)).
A number sign (#) is used with this entry because of evidence that narcolepsy-7 (NRCLP7) is caused by heterozygous mutation in the MOG gene (159465) on chromosome 6p22. One such family has been reported. For a phenotypic description and a discussion of genetic heterogeneity of narcolepsy, see 161400. Clinical Features Hor et al. (2011) reported a large 4-generation family in which 12 living individuals had narcolepsy and cataplexy. A thirteenth had evidence suggesting the diagnosis. Seven affected individuals were obese and 4 had type 2 diabetes. Hypocretin (HCRT; 602358) levels in cerebrospinal fluid were undetectable in those tested.
For a phenotypic description and a discussion of genetic heterogeneity of narcolepsy, see 161400. Mapping Kornum et al. (2011) reported genomewide association analyses for narcolepsy with replication and fine mapping across 3 ethnic groups (3,406 individuals of European ancestry, 2,414 Asians, and 302 African Americans) and identified a single-nucleotide polymorphism (SNP) in a 3-prime untranslated region of the purinergic receptor subtype P2Y11 gene (P2RY11) on chromosome 19p13.2 associated with narcolepsy (rs2305795, combined p = 6.1 x 10(-10), OR = 1.28, 95% CI 1.19-1.39, n = 5689). The disease-associated allele was correlated with reduced expression of P2RY11 in CD8+ T lymphocytes (339% reduced, p = 0.003) and natural killer cells (P = 0.031), but not in other peripheral blood mononuclear cell types. The low-expression variant was also associated with reduced P2RY11-mediated resistance to ATP-induced cell death in T lymphocytes (p = 0.0007) and natural killer cells (P = 0.001). Kornum et al. (2011) concluded that their results identified P2RY11 as an important regulator of immune cell survival, with possible implications in narcolepsy and other autoimmune diseases.
Overview Narcolepsy is a sleep disorder that makes people very drowsy during the day. People with narcolepsy find it hard to stay awake for long periods of time. They fall asleep suddenly. This can cause serious problems in their daily routine. Sometimes narcolepsy also causes a sudden loss of muscle tone, known as cataplexy (KAT-uh-plek-see). This can be triggered by strong emotion, especially laughter. Narcolepsy is divided into two types.
For a phenotypic description and a discussion of genetic heterogeneity of narcolepsy, see 161400. Mapping Dauvilliers et al. (2004) reported a large French family in which 4 members had narcolepsy-cataplexy and 10 others had a milder form with isolated recurrent naps or lapses into sleep. The disorder was inherited in an autosomal dominant pattern. Genomewide linkage analysis identified a 5.15-Mb (12.6-cM) candidate region between markers D21S267 and ABCG1 on chromosome 21q, yielding a maximum 2-point lod score of 3.36 at D21S1245 and a maximum multipoint parametric lod score of 4.00 at 21GT26K. Two patients with the more severe form and 6 patients with the milder form were DQB1*0602-positive. A shared haplotype was observed in all affected individuals. Molecular analysis of the PEP19 gene (601629) revealed no mutations.
Cameron and Sinclair (1997) stated that 14 heterozygous mutations in SOX9 and 10 translocations involving 17q have been described in 28 patients with campomelic dysplasia.
Campomelic dysplasia is a rare genetic disorder that affects the development of the skeleton, reproductive system, and face. Symptoms of campomelic dysplasia may include bowing of the legs, dislocated hips, small lungs and chest, and external genitalia that do not look clearly male or clearly female (ambiguous genitalia). In addition, infants with campomelic dysplasia have distinctive facial features including a small chin with cleft palate, prominent eyes, flat face, and a large head. Many infants die at an early age due to breathing problems. Campomelic dysplasia usually results from a new genetic change ( DNA variant) in or the near the SOX9 gene. Diagnosis is based on physical findings and x-ray (radiograph) findings and may be confirmed by genetic testing.
Summary Clinical characteristics. Campomelic dysplasia (CD) is a skeletal dysplasia characterized by distinctive facies, Pierre Robin sequence with cleft palate, shortening and bowing of long bones, and clubfeet. Other findings include laryngotracheomalacia with respiratory compromise and ambiguous genitalia or normal female external genitalia in most individuals with a 46,XY karyotype. Many affected infants die in the neonatal period; additional problems identified in long-term survivors include short stature, cervical spine instability with cord compression, progressive scoliosis, and hearing impairment. Diagnosis/testing. The diagnosis of CD is usually based on clinical and radiographic findings. Molecular genetic testing of SOX9 , the only gene in which pathogenic variants are known to cause CD, detects pathogenic variants or chromosome rearrangements in approximately 95% of affected individuals.
Clinical Features In a 5-generation family with multiple musculoskeletal anomalies, Stalker and Zori (1997) described an apparently new malformation syndrome with autosomal dominant inheritance. The features included the Pierre Robin-type cleft palate (261800), pectus excavatum, rib anomalies, and hypoplasia of the distal segments of the scapulae. The propositus at birth was mildly hypotonic with a Robin-type cleft involving the soft and hard palate with micrognathia, and with moderately severe pectus excavatum. Radiographs showed 11 pairs of ribs and hypoplasia of the inferior subscapular area, more pronounced on the left than on the right. The mother at birth had a U-shaped cleft of the soft palate and micrognathia, a grade II/VI systolic murmur that resolved neonatally, moderate pectus excavatum, low-set hairline, and hyperextensibility of joints.
Campomelic dysplasia is a severe disorder that affects development of the skeleton, reproductive system, and other parts of the body. This condition is often life-threatening in the newborn period. The term "campomelic" comes from the Greek words for "bent limb." Affected individuals are typically born with bowing of the long bones in the legs, and occasionally, bowing in the arms. Bowing can cause characteristic skin dimples to form over the curved bone, especially on the lower legs. People with campomelic dysplasia usually have short legs, dislocated hips, underdeveloped shoulder blades, 11 pairs of ribs instead of 12, bone abnormalities in the neck, and inward- and upward-turning feet (clubfeet ).
A rare skeletal dysplasia characterized by peculiar facial anomalies, Pierre Robin sequence, cleft palate, shortening and bowing of long bones. Sexual ambiguity or female external genitalia is possible individuals with a male karyotype. Epidemiology Prevalence at birth is approximately 1/40,000-80,000. Clinical description The clinical features include a relatively large head, Pierre Robin sequence with cleft palate, flat face, laryngotracheomalacia, respiratory distress, 11 pairs of ribs, ambiguous genitalia or normal female external genitalia in an individual with a 46,XY karyotype. It also includes dislocated hips, short bowed femura and tibiae (lower limbs more frequently than upper limbs), pretibial skin dimples (bowing of the lower leg is often associated with a skin dimple over the apex of curve) and clubfeet. A few cases of a variant syndrome, referred to as ``acampomelic campomelic dysplasia'' have been described.
Campomelic dysplasia This condition is inherited in an autosomal dominant manner Specialty Medical genetics Campomelic dysplasia ( CMD ) is a rare genetic disorder characterized by bowing of the long bones and many other skeletal and extraskeletal features. It is frequently lethal in the neonatal period due to respiratory insufficiency , but the severity of the disease is variable, and some patients survive into adulthood. The name is derived from the Greek roots campo (or campto ), meaning bent, and melia , meaning limb. An unusual aspect of the disease is that up to two-thirds of affected 46,XY genotypic males display a range of disorders of sexual development (DSD) and genital ambiguities or may even develop as normal phenotypic females as in complete 46 XY sex reversal . An atypical form of the disease with absence of bowed limbs is called, prosaically, acampomelic campomelic dysplasia (ACD) and is found in about 10% of patients, particularly those surviving the neonatal period. [ citation needed ] Contents 1 Signs and symptoms 2 Genetics 3 Diagnosis 3.1 Screening 4 Prognosis 5 Epidemiology 6 References 7 External links Signs and symptoms [ edit ] While the definitive presentation of the disease is a patient having bowed lower limbs and sex reversal in 46,XY males, there are other clinical criteria that can be used, absent these characteristics, to make the diagnosis.
Khajavi et al. (1976) recognized three varieties of campomelic syndrome: (1) the long-limb form, in which the bent bones are of normal width and only slightly shortened and the arms are rarely involved; (2) the short-limbed form, in which the bent bones are short and wide; and (3) a short-limbed form with associated cloverleaf skull deformity ('Kleeblattschaedel'). Both the long-bone and the short-bone forms may be recessive. Krous et al. (1979) raised the question of intrauterine viral infection in 2 infants with campomelia. Both showed hydrocephalus and hydromyelia and the placenta in one showed focal proliferative villitis. Mellows et al. (1980) described XX female sibs with camptomelic syndrome of the long-limbed variety. Both infants died soon after birth. Fryns et al. (1981) achieved prenatal diagnosis by ultrasonography in a woman who had delivered an infant with presumed camptomelic dwarfism (although no diagnostic studies were performed and the infant lived only a few minutes).
Using genetic engineering in attempts to produce babies free of mitochondrial disease is controversial in some circles and raises important ethical issues . [26] [27] A male baby was born in Mexico in 2016 from a mother with Leigh syndrome using spindle transfer. [28] In September 2012 a public consultation was launched in the UK to explore the ethical issues involved. [29] Human genetic engineering was used on a small scale to allow infertile women with genetic defects in their mitochondria to have children. [30] In June 2013, the United Kingdom government agreed to develop legislation that would legalize the 'three-person IVF ' procedure as a treatment to fix or eliminate mitochondrial diseases that are passed on from mother to child.