Healthcare workers could possibly spread the bacteria or contaminate surfaces through hand contact. [25] The rate of C. difficile acquisition is estimated to be 13% in those with hospital stays of up to two weeks, and 50% with stays longer than four weeks. [26] Long-term hospitalization or residence in a nursing home within the previous year are independent risk factors for increased colonization . [27] Acid suppression medication [ edit ] Increasing rates of community-acquired CDI are associated with the use of medication to suppress gastric acid production: H2-receptor antagonists increased the risk 1.5-fold, and proton pump inhibitors by 1.7 with once-daily use and 2.4 with more than once-daily use. [28] [29] Diarrheal illnesses [ edit ] People with a recent history of diarrheal illness are at increased risk of becoming colonized by C. difficile when exposed to spores, including laxative abuse and gastrointestinal pathogens. [10] Disturbances that increase intestinal motility are thought to transiently elevate the concentration of available dietary sugars, allowing C. difficile to proliferate and gain a foothold in the gut. [30] Although not all colonization events lead to disease, asymptomatic carriers remain colonized for years at a time. [10] During this time, the abundance of C. difficile varies considerably day-to-day, causing periods of increased shedding that could substantially contribute to community-acquired infection rates. [10] Other [ edit ] As a result of suppression of healthy bacteria, via a loss of bacterial food source, prolonged use of an elemental diet increases the risk of developing C. difficile infection. [31] Low serum albumin levels is a risk factor for the development of C. difficile infection and when infected for severe disease. [32] [33] The protective effects of serum albumin may be related to the capability of this protein to bind C. difficile toxin A and toxin B, thus impairing entry into enterocytes. [33] Pathophysiology [ edit ] The use of systemic antibiotics, including broad-spectrum penicillins/cephalosporins, fluoroquinolones, and clindamycin, causes the normal microbiota of the bowel to be altered. ... Two years later, the same strain of the bacterium was detected in New Zealand. [111] On 28 May 2011, an outbreak in Ontario had been reported, with 26 fatalities as of 24 July 2011. [112] In 2012/2013, a total of 27 people at one hospital in the south of Sweden (Ystad) were infected with 10 deaths.
Cognitive and associative effects of CBD are also hard to treat as we are still unsure of many of the treatments for the symptomatic diseases that ensue like dementia, aphasia, neglect, apraxia and others. [ citation needed ] Epidemiology [ edit ] Clinical presentation of CBD usually does not occur until age 60, with the earliest recorded diagnosis and subsequent postmortem verification being age 28. [19] Although men and women present with the disease, some analysis has shown a predominant appearance of CBD in women.
Corticobasal degeneration is characterized by the break down (degeneration) of parts of the brain, including the cerebral cortex and basal ganglia. The cerebral cortex is responsible for most of the brain's processing of information, and the basal ganglia are deep brain structures that help start and control movement. Signs and symptoms of corticobasal degeneration include poor coordination, loss of movement, rigidity, poor balance, unnatural posturing of the muscles, intellectual (cognitive) impairment, speech impairment, muscular jerks, and difficulty swallowing. These symptoms develop and worsen over time. Currently the cause of corticobasal degeneration is not known. Treatment depends on the symptoms in each person. People with corticobasal degeneration usually do not survive beyond an average of 7 years after symptoms begin.
A rare neurologic disease characterized by multifaceted motor system dysfunctions and cognitive defects such as asymmetric rigidity, bradykinesia, limb apraxia, and visuospatial dysfunction. Epidemiology The prevalence of Corticobasal syndrome (CBS) is unknown. Clinical description The disease shows a wide clinical variability between patients with many developing a relatively pure motor syndrome, and others displaying a combination of motor and cognitive deficits. Disease onset is insidious and usually occurs in the 6th to 7th decade of life with symptoms typically being unilateral at first, and the arm being more commonly affected than the leg. It may begin primarily as a movement disorder with rigidity, bradykinesia and tremor, in association with frontal signs, cortical sensory loss, alien limb phenomenon, stimulus induced myoclonus and progressive limb apraxia, which may become bilateral, though usually asymmetrical, as the disease progresses.
Corticobasal syndrome (CBS) is a rare, progressive atypical Parkinsonism syndrome and is a tauopathy related to frontotemporal dementia . [1] [2] CBS is typically caused by the deposit of tau proteins forming in different areas of the brain. [1] [3] Contents 1 Classification 2 Symptoms 3 Pathophysiology 4 Diagnosis 4.1 Differential 5 Prognosis 6 References Classification [ edit ] CBS is the most common type of corticobasal degeneration (CBD) although the terms CBD and CBS have been used interchangeably in the past. [2] The other three phenotypes of CBD are: frontal-behavioral dysexecutive-spatial syndrome (FBS) nonfluent/agrammatic variant of primary progressive aphasia (naPPA), and progressive supranuclear palsy syndrome (PSPS). [1] [4] Symptoms [ edit ] Symptoms of CBS include apraxia , alien limb phenomenon, frontal deficits, and extrapyramidal motor symptoms such as myoclonus or rigidity. [5] Movement deficits often begin on one side and progress to the other. [1] Pathophysiology [ edit ] CBD is the pathology underlying approximately 50% of CBS cases. [6] Diagnosis [ edit ] The Armstrong criteria were proposed in 2013; the accuracy of these is limited and further research is needed. [7] Symptoms may be symmetric or asymmetric, with one or more of the following: limb rigidity or akinesia limb dystonia limb myoclonus, plus one of: orobuccal or limb apraxia cortical sensory deficit alien limb phenomena (more than simple levitation) The onset is insidious with gradual progression, lasting one year or more, with no exclusion criteria present. The diagnosis is more likely if onset is after age 50, there is no family history (2 or more relatives), [ clarification needed ] and there is no genetic mutation affecting T [ clarification needed ] (e.g., MAPT). [8] Probably sporadic CBS is more likely if there are two of: limb rigidity or akinesia limb dystonia limb myoclonus plus two of: orobuccal or limb apraxia, cortical sensory deficit alien limb phenomena (more than simple levitation) [8] The diagnosis is excluded if there is evidence of: Lewy body disease multiple system atrophy Alzheimer's disease amyotrophic lateral sclerosis semantic or logopenic variant primary progressive aphasia structural lesion suggestive of focal cause granulin mutation or reduced plasma progranulin levels TDP-43 or fused in sarcoma (FUS) mutations [8] The diagnostic criteria for clinical use may result in a misdiagnosis of other tau-based diseases. [7] The probable criteria are proposed for clinical research. [7] Differential [ edit ] Other degenerative pathologies that can cause corticobasal syndrome include: Alzheimer's disease Pick's disease with Pick bodies Lewy body dementias Neurofilament inclusion body disease Creutzfeldt-Jakob disease Frontotemporal degeneration due to progranulin gene mutation Motor neuron disease‐inclusion dementia. [9] The symptoms of classic CBS differ from CBD in that CBD also includes cognitive deficits in the executive functions . [10] Prognosis [ edit ] The average survival time after disease onset is estimated at 6.5 years. [2] References [ edit ] ^ a b c d Parmera JB, Rodriguez RD, Neto AS, Nitrini R, Brucki SM (2016). "Corticobasal syndrome: A diagnostic conundrum" . Dement Neuropsychol (Review). 10 (4): 267–75. doi : 10.1590/s1980-5764-2016dn1004003 . PMC 5619264 . PMID 29213468 . ^ a b c Constantinides VC, Paraskevas GP, Paraskevas PG, Stefanis L, Kapaki E (August 2019). "Corticobasal degeneration and corticobasal syndrome: A review" . Clinical Parkinsonism & Related Disorders . 1 : 66–71. doi : 10.1016/j.prdoa.2019.08.005 .
Adults with dyslexia can often read with good comprehension, though they tend to read more slowly than others without a learning difficulty and perform worse in spelling tests or when reading nonsense words–a measure of phonological awareness. [22] Associated conditions Dyslexia often co-occurs with other learning disorders, but the reasons for this comorbidity have not been clearly identified. [23] These associated disabilities include: Dysgraphia : A disorder involving difficulties with writing or typing, sometimes due to problems with eye–hand coordination ; it also can impede direction- or sequence-oriented processes, such as tying knots or carrying out repetitive tasks. [24] In dyslexia, dysgraphia is often multifactorial, due to impaired letter-writing automaticity , organizational and elaborative difficulties, and impaired visual word forming, which makes it more difficult to retrieve the visual picture of words required for spelling. [24] Attention deficit hyperactivity disorder (ADHD): A disorder characterized by problems sustaining attention, hyperactivity, or acting impulsively. [25] Dyslexia and ADHD commonly occur together. [5] [26] [27] Approximately 15% [11] or 12–24% of people with dyslexia have ADHD; [28] and up to 35% of people with ADHD have dyslexia. [11] Auditory processing disorder : A listening disorder that affects the ability to process auditory information. [29] [30] This can lead to problems with auditory memory and auditory sequencing . ... S2CID 7433822 . ^ Peterson, Robin L.; Pennington, Bruce F. (28 March 2015). "Developmental Dyslexia".
Overview Dyslexia is a learning disorder that involves difficulty reading due to problems identifying speech sounds and learning how they relate to letters and words (decoding). Also called a reading disability, dyslexia is a result of individual differences in areas of the brain that process language. Dyslexia is not due to problems with intelligence, hearing or vision. Most children with dyslexia can succeed in school with tutoring or a specialized education program. Emotional support also plays an important role. Though there's no cure for dyslexia, early assessment and intervention result in the best outcome.
A little over 50% of all people with Down syndrome suffer from obstructive sleep apnea, [28] and some physicians advocate routine testing of this group. [29] In other craniofacial syndromes, the abnormal feature may actually improve the airway, but its correction may put the person at risk for obstructive sleep apnea after surgery when it is modified.
Description Obstructive sleep apnea is a common, chronic, complex disease associated with serious cardiovascular and neuropsychologic sequelae and with substantial social and economic costs (Palmer et al., 2003). Clinical Features Strohl et al. (1978) described 2 males and their father with severe hypersomnolence and obstructive sleep apnea. A third son, although asymptomatic, was shown to have upper-airway obstruction during sleep. Electromyographic recordings of genioglossal muscle activity showed loss of tonic activity in early stages of sleep when sleep apnea occurred. (The bilateral genioglossus muscles play a crucial role in the normal mechanism for maintaining a patent oropharyngeal lumen, especially during sleep in the supine position, for they are the muscles that force the tongue forward during inspiration.)
Overview Obstructive sleep apnea is the most common sleep-related breathing disorder. People with obstructive sleep apnea repeatedly stop and start breathing while they sleep. There are several types of sleep apnea. Obstructive sleep apnea occurs when the throat muscles relax and block the airway. This happens off and on many times during sleep. A sign of obstructive sleep apnea is snoring. Treatments for obstructive sleep apnea are available. One treatment is a device that uses positive pressure to keep the airway open during sleep.
Obstructive sleep apnea is a condition in which individuals experience pauses in breathing (apnea) during sleep, which are associated with partial or complete closure of the throat (pper airway). Complete closure can lead to apnea while partial closure allows breathing but decrease the intake of oxygen (hypopnea). Individuals with obstructive sleep apnea may experience interrupted sleep with frequent awakenings and loud snoring. Repeated pauses in breathing lead to episodes of lower-than-normal oxygen levels (hypoxemia) and a buildup of carbon dioxide (hypercapnia) in the bloodstream. Interrupted and poor quality sleep can lead to daytime sleepiness and fatigue, impaired attention and memory, headaches, depression, and sexual dysfunction.
This demonstrates that students who acknowledge their academic limitations but are also aware of their potential to succeed in other intellectual tasks see themselves as intellectually competent individuals, which increases their self-esteem. [28] Research involving individuals with learning disabilities who exhibit challenging behaviors who are subsequently treated with antipsychotic medications provides little evidence that any benefits outweigh the risk. [29] Causes [ edit ] The causes for learning disabilities are not well understood, and sometimes there is no apparent cause for a learning disability. ... LDAO – North Bay and Area News Release . Retrieved 28 April 2015 . ^ Gates, Bob & Mafuba, Kay.
GHB accumulates in the nervous system and can cause ataxia as well as other neurological dysfunction. [26] Wilson's disease [ edit ] Wilson's disease is an autosomal - recessive gene disorder whereby an alteration of the ATP7B gene results in an inability to properly excrete copper from the body. [27] Copper accumulates in the nervous system and liver and can cause ataxia as well as other neurological and organ impairments. [28] Gluten ataxia [ edit ] Play media A male with gluten ataxia: previous situation and evolution after three months of a gluten-free diet Gluten ataxia is an autoimmune disease triggered by the ingestion of gluten . [29] [30] Early diagnosis and treatment with a gluten-free diet can improve ataxia and prevent its progression.
This suggests growth deficiency may be less critical for understanding the disabilities of FASD than the neurobehavioral sequelae to the brain damage. [19] Facial features [ edit ] Several characteristic craniofacial abnormalities are often visible in individuals with FAS. [28] The presence of FAS facial features indicates brain damage , although brain damage may also exist in their absence.
T1-weighted imaging may identify scarring of cardiac tissues while T2-weighted imaging may identify oedema and inflammation of cardiac tissue which is associated with acute clinical signs of chest pain and fainting episodes. [27] Pulsus bisferiens may occasional be found during examination. [28] Obstructive or non-obstructive [ edit ] Depending on whether the distortion of normal heart anatomy causes an obstruction of the outflow of blood from the left ventricle of the heart, HCM can be classified as obstructive or non-obstructive. ... June 22, 2016. Archived from the original on 28 July 2016 . Retrieved 31 August 2016 . ^ a b c "What Are the Signs and Symptoms of Cardiomyopathy?" ... NHLBI . 22 June 2016. Archived from the original on 28 July 2016 . Retrieved 10 November 2017 . ^ a b c "What Is Cardiomyopathy?"
Familial hypertrophic cardiomyopathy (HCM) is an inherited heart condition characterized by thickening of the heart muscle. The thickening most often occurs in the muscle wall that separates the left and right ventricles from each other (interventricular septum). This may restrict the flow of oxygen-rich blood from the heart, or it may lead to less efficient pumping of blood. Signs and symptoms can vary. While some people have no symptoms, others may have chest pain, shortness of breath, palpitations, lightheadedness, dizziness, and/or fainting. Even in the absence of symptoms, familial HCM can have serious consequences such as life-threatening arrhythmias , heart failure , and an increased risk of sudden death.
The theater group has put on several productions and appeared both locally in Israel and abroad in London and Broadway. [28] Katie Kelly , a gold medal winning paraolympian.
A rare ciliopathy characterized by progressive hearing and visual loss in the first decades of life and, in some cases, vestibular dysfunction. Patients have normal hearing at birth. Onset of hearing loss is usually in late childhood or adolescence after development of speech. Profound deafness is mostly reported by middle age. Retinitis pigmentosa related visual loss also develops in late childhood or adolescence. Developmental motor milestones are generally normal but vestibular dysfunction may occur in adulthood.
Usher syndrome is a genetic disorder characterized by sensorineural hearing loss or deafness and progressive vision loss due to retinitis pigmentosa . Sensorineural hearing means it is caused by abnormalities of the inner ear . Retinitis pigmentosa is an eye disease that affects the layer of light-sensitive tissue at the back of the eye ( the retina ). Vision loss occurs as the light-sensing cells of the retina gradually deteriorate. Night vision loss begins first, followed by blind spots that develop in the side (peripheral) vision, that can enlarge and merge to produce tunnel vision (loss of all peripheral vision).
The latter finding reflects the increased lung stiffness (reduced lung compliance) associated with pulmonary fibrosis, which leads to increased lung elastic recoil. [28] Measurement of static lung volumes using body plethysmography or other techniques typically reveals reduced lung volumes (restriction). ... IPF was the most common diagnosis (28%) followed by connective tissue disease-related ILD (14%), hypersensitivity pneumonitis (7%) and non-specific interstitial pneumonia (NSIP) (7%).
An interstitial lung disease with a poor prognosis, that is characterized by the progressive formation of scar tissue within the lungs in the absence of any known cause. Epidemiology Idiopathic pulmonary fibrosis (IPF) incidence appears to be increasing. Reported incidences range from 0.2 per 100.000 per year to 94 per 100.000 per year. The prevalence is estimated to be higher in men than in women. Clinical description The mean age at presentation is 66 years. IPF initially usually manifests with symptoms of breathlessness on exertion and dry coughing.
Denborough et al. (1982) found susceptibility to malignant hyperpyrexia and accompanying muscle abnormalities in 5 of 15 parents whose children had died of sudden infant death syndrome (SIDS). A 28-year-old man, whose son had died of SIDS at age 16 months, had had 3 cardiac arrests after appendectomy at age 19 and his mother had severe hyperpyrexia after hysterectomy.
The King-Denborough syndrome (KDS) is a congenital myopathy associated with susceptibility to malignant hyperthermia , skeletal abnormalities and dysmorphic features with characteristic facial appearance. Although the cause of King-Denborough syndrome is not fully understood, at least some cases have been attributed to the ryanodine receptor gene (RYR1), which has been tied to malignant hyperthermia and central core disease .
King-Denborough syndrome is a rare genetic non-dystrophic myopathy characterized by the triad of congenital myopathy, dysmorphic features and susceptibility to malignant hyperthermia. Patients present with a wide phenotypic range, including delayed motor development, muscle weakness and fatigability, ptosis and facies myopathica (with or without creatine kinase elevations), skeletal abnormalities (e.g. short stature, scoliosis, kyphosis, lumbar lordosis and pectus carinatum/excavatum), mild dysmorphic facial features (e.g. hypertelorism, down-slanting palpebral fissures, epicanthic folds, low set ears, micrognathia), webbing of the neck, cryptorchidism, and a susceptibility to malignant hyperthermia and/or rhabdomyolysis due to intensive physical strain, viral infection or statin use.
The case-control study showed an increased frequency in Graves disease, compared to controls, of DRB1*0304 (47% vs 24%; pc less than 1.4 x 10-5), DQB1*02 (58% vs 46%; pc less than 0.035), DQB1*0301/4 (42% vs 28%; pc less than 3.5 x 10-3) and DQA1*0501 (67% vs 39%; pc less than 7 x 10-6).
Graves disease is a condition that affects the function of the thyroid , which is a butterfly-shaped gland in the lower neck. The thyroid makes hormones that help regulate a wide variety of critical body functions. For example, thyroid hormones influence growth and development, body temperature, heart rate, menstrual cycles, and weight. In people with Graves disease, the thyroid is overactive and makes more hormones than the body needs. The condition usually appears in mid-adulthood, although it may occur at any age.
Risch et al. (1989, 1990) ascertained 43 Ashkenazi Jewish probands with idiopathic torsion dystonia with onset before age 28 years and studied all available first- and second-degree relatives.
Early-onset primary dystonia is a condition characterized by progressive problems with movement, typically beginning in childhood. Dystonia is a movement disorder that involves involuntary tensing of the muscles (muscle contractions), twisting of specific body parts such as an arm or a leg, rhythmic shaking (tremors), and other uncontrolled movements. A primary dystonia is one that occurs without other neurological symptoms, such as seizures or a loss of intellectual function (dementia). Early-onset primary dystonia does not affect a person's intelligence. On average, the signs and symptoms of early-onset primary dystonia appear around age 12.
Early-onset generalized dystonia is a neurologic movement disorder that usually begins in childhood or adolescence. This is the most common hereditary form of dystonia. Symptoms start in one part of the body (usually an arm, foot, or leg) and are usually first apparent with actions such as writing or walking. With time, the contractions may spread to other parts of the body, causing the muscles to twist the body into unnatural positions. Symptoms can vary greatly, even among members of the same family. For some, the disorder can cause significant disability, while others may experiences only isolated writer’s cramp. A small deletion in the DYT1 gene is the major cause of early-onset dystonia.
Mostofsky et al. (1996) reported a father and his daughter with torsion dystonia that could not be attributed to exogenous factors or other neurologic disorders. The first signs of the disorder in both patients appeared during the first year of life. The main manifestations included generalized dystonia with severe involvement of the legs and mild involvement of the face and arms, no progression of symptoms after 10 years of age, no evidence of parkinsonism, and no intellectual, cerebellar, or sensory involvement. Among 850 cases of idiopathic torsion dystonia (128100) collected from the literature, the authors found only 2 reports with onset before 3 years of age; the inheritance pattern in these 2 cases was not reported. No response to dopaminergic agents excluded dopa-responsive dystonia (128230).
Lichen planopilaris is considered an orphan disease with no definitive prevalence data and no proven effective treatments. [25] [26] Other variants may include: Lichen planus pemphigoides characterized by the development of tense blisters atop lesions of lichen planus or the development vesicles de novo on uninvolved skin. [27] Keratosis lichenoides chronica (also known as "Nekam's disease") is a rare dermatosis characterized by violaceous papular and nodular lesions, often arranged in a linear or reticulate pattern on the dorsal hands and feet, extremities, and buttock, and some cases manifest by sorrheic dermatitis-like eruption on the scalp and face; also palmo plantar keratosis has been reported. [16] [28] [29] Lichenoid keratoses (also known as "benign lichenoid keratosis," and "Solitary lichen planus" [16] ) is a cutaneous condition characterized by brown to red scaling maculopapules, found on sun-exposed skin of extremities. [16] [30] Restated, this is a cutaneous condition usually characterized by a solitary dusky-red to violaceous papular skin lesion. [31] Lichenoid dermatitis represents a wide range of cutaneous disorders characterized by lichen planus-like skin lesions. [16] [30] Mucous membranes [ edit ] Lichen planus on the lips and the lateral border of the tongue Lichen planus affecting mucosal surfaces may have one lesion or be multifocal. [32] Examples of lichen planus affecting mucosal surfaces include: [32] Esophageal lichen planus , affecting the esophageal mucosa. ... "Pathophysiology, etiologic factors, and clinical management of oral lichen planus, part I: facts and controversies". Clinics in Dermatology . 28 (1): 100–8. doi : 10.1016/j.clindermatol.2009.03.004 . PMID 20082959 . ^ a b c d e f g h i j k Lodi, Giovanni; Manfredi, Maddalena; Mercadante, Valeria; Murphy, Ruth; Carrozzo, Marco (28 February 2020). "Interventions for treating oral lichen planus: corticosteroid therapies" .
Lichen planus (LP) is a common inflammatory dermatosis characterized by the development of pruritic violaceous papules or plaques on mucocutaneous surfaces. Eruptions can involve the face, neck, limbs, back, genitalia, tongue, buccal mucosa, nails, and scalp. LP comprises rare variants affecting the skin and the mucosa. Rare cutaneous LP includes linear LP (referring to blaschkoid and zosteriform distributions of lichenoid lesions), actinic LP, annular LP, atrophic LP, annular atrophic LP, lichen planopilaris (comprising Graham Little-Piccardi-Lassueur syndrome and frontal fibrosing alopecia), lichen planus pigmentosus, and lichen planus pemphigoides (see these terms). Rare mucosal LP includes vulvovaginal gingival syndrome and LP sialadenitis (see these terms).
Rare lichen planus (rare LP) refers to several rare variants of lichen planus , which is a condition that affects the skin and/or mouth. The signs and symptoms vary by subtype but generally include an itchy rash . In most cases, the exact underlying cause of rare LP is unknown; however, studies suggest that it may be due to an allergic or immune reaction . Treatment is based on the signs and symptoms present in each person; however, intervention is not always necessary as some cases of rare LP resolve on their own.
Overview Lichen planus (LIE-kun PLAY-nus) is a condition of the skin, hair, nails, mouth and genitals. On skin, lichen planus often appears as purple, itchy, flat bumps that develop over several weeks. In the mouth and genital mucosa, lichen planus forms lacy white patches, sometimes with painful sores. Mild lichen planus of the skin may not need treatment. If the condition causes pain or intense itching, you may need prescription medicine. Symptoms Symptoms of lichen planus vary depending on the part of the body affected.
Addressing these factors has been found to improve comfort in some studies. [27] A 2010 survey by NIOSH showed that 2/3 of the 5 million carpal tunnel cases in the US that year were related to work. [28] Women have more work-related carpal tunnel syndrome than men. [29] Speculation that CTS is work-related is based on claims such as CTS being found mostly in the working adult population, though evidence is lacking for this. ... National Institute of Neurological Disorders and Stroke . January 28, 2016. Archived from the original on 3 March 2016 . ... National Institute of Neurological Disorders and Stroke . December 28, 2010. Archived from the original on December 22, 2010. ^ Netter, Frank (2011).
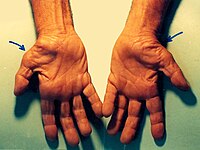
Carpal tunnel syndrome is a disorder caused by disturbances in nerve function (neuropathy), leading to pain and numbness or tingling (paresthesia) primarily in the wrist and hand. While carpal tunnel syndrome can occur at any age, it most often affects people between the ages of 40 and 60. In more than half of cases, both hands are affected; however, the severity may vary between hands. When only one hand is affected, it is most often the hand used for writing (the dominant hand). In carpal tunnel syndrome, the pain or paresthesia is usually felt in the wrist, the palm, and the first four fingers of the hand.
Overview Carpal tunnel syndrome is caused by pressure on the median nerve. The carpal tunnel is a narrow passageway surrounded by bones and ligaments on the palm side of the hand. When the median nerve is compressed, symptoms can include numbness, tingling, and weakness in the hand and arm. The anatomy of the wrist, health problems and possibly repetitive hand motions can contribute to carpal tunnel syndrome. Proper treatment usually relieves the tingling and numbness and restores wrist and hand function.
A number sign (#) is used with this entry because at least some cases of carpal tunnel syndrome are caused by heterozygous mutation in the TTR gene, encoding transthyretin (176300), on chromosome 18q12. Susceptibility to the development of carpal tunnel syndrome (613353) may also be conferred by heterozygous mutation in the SH3TC2 gene (608206) on chromosome 5q32. Clinical Features Danta (1975) reported carpal tunnel syndrome (constrictive median neuropathy) in 4 persons in 3 generations with male-to-male transmission. Symptoms began in the first decade in father and son, and in both the median nerve at operation was found to be constricted under a thickened transverse carpal ligament. Carpal tunnel syndrome has been described in amyloid neuropathy (see 176300) and in mucopolysaccharidoses (e.g., 253200) and mucolipidoses (252600).
Some neurochemical abnormalities that occur in fibromyalgia also regulate mood, sleep, and energy, thus explaining why mood, sleep, and fatigue problems are commonly co-morbid with fibromyalgia. [15] Genetics [ edit ] A mode of inheritance is currently unknown, but it is most probably polygenic . [8] Research has also demonstrated that fibromyalgia is potentially associated with polymorphisms of genes in the serotoninergic , [25] dopaminergic [26] and catecholaminergic systems. [27] However, these polymorphisms are not specific for fibromyalgia and are associated with a variety of allied disorders (e.g. chronic fatigue syndrome , [28] irritable bowel syndrome [29] ) and with depression. [30] Individuals with the 5-HT2A receptor 102T/C polymorphism have been found to be at increased risk of developing fibromyalgia. [31] Lifestyle and trauma [ edit ] Stress may be an important precipitating factor in the development of fibromyalgia. [32] Fibromyalgia is frequently comorbid with stress-related disorders such as chronic fatigue syndrome , posttraumatic stress disorder , irritable bowel syndrome and depression. [33] A systematic review found significant association between fibromyalgia and physical and sexual abuse in both childhood and adulthood, although the quality of studies was poor. [34] Poor lifestyles including being a smoker, obesity and inactivity may increase the risk of an individual developing fibromyalgia. [35] A meta-analysis found psychological trauma to be associated with fibromyalgia, although not as strongly as in chronic fatigue syndrome . [36] Some authors have proposed that, because exposure to stressful conditions can alter the function of the hypothalamic-pituitary-adrenal (HPA) axis , the development of fibromyalgia may stem from stress-induced disruption of the HPA axis. [37] Sleep disturbances [ edit ] Impaired sleep is a risk factor for fibromyalgia. [4] In 1975, Moldofsky and colleagues reported the presence of anomalous alpha wave activity (typically associated with arousal states) measured by electroencephalogram (EEG) during non- rapid eye movement sleep of "fibrositis syndrome". [18] By disrupting stage IV sleep consistently in young, healthy subjects, the researchers reproduced a significant increase in muscle tenderness similar to that experienced in "neurasthenic musculoskeletal pain syndrome" but which resolved when the subjects were able to resume their normal sleep patterns. [38] Mork and Nielsen used prospective data and identified a dose-dependent association between sleep problems and risk of fibromyalgia. [39] Psychological factors [ edit ] There is strong evidence that major depression is associated with fibromyalgia as with other chronic pain conditions (1999), [40] although the direction of the causal relationship is unclear. [41] A comprehensive review into the relationship between fibromyalgia and major depressive disorder (MDD) found substantial similarities in neuroendocrine abnormalities, psychological characteristics, physical symptoms and treatments between fibromyalgia and MDD, but currently available findings do not support the assumption that MDD and fibromyalgia refer to the same underlying construct or can be seen as subsidiaries of one disease concept. [42] Indeed, the sensation of pain has at least two dimensions: a sensory dimension which processes the magnitude and location of the pain, and an affective-motivational dimension which processes the unpleasantness.
Overview Fibromyalgia is a disorder characterized by widespread musculoskeletal pain accompanied by fatigue, sleep, memory and mood issues. Researchers believe that fibromyalgia amplifies painful sensations by affecting the way your brain and spinal cord process painful and nonpainful signals. Symptoms often begin after an event, such as physical trauma, surgery, infection or significant psychological stress. In other cases, symptoms gradually accumulate over time with no single triggering event. Women are more likely to develop fibromyalgia than are men. Many people who have fibromyalgia also have tension headaches, temporomandibular joint (TMJ) disorders, irritable bowel syndrome, anxiety and depression.
Fibromyalgia is a common condition characterized by long-lasting (chronic) pain affecting many areas of the body. The pain is associated with tenderness that occurs with touch or pressure on the muscles, joints, or skin. Some affected individuals also report numbness, tingling, or a burning sensation (paresthesia) in the arms and legs. Other signs and symptoms of fibromyalgia include excessive tiredness (exhaustion); sleep problems, such as waking up feeling unrefreshed; and problems with memory or thinking clearly. People with fibromyalgia often report additional types of pain, including headaches, back and neck pain, sore throat, pain or clicking in the jaw (temporomandibular joint dysfunction), and stomach pain or digestive disorders such as irritable bowel syndrome.
Causes or triggers include: [24] Falling asleep and waking: hypnagogic and hypnopompic hallucinations, which are entirely normal [25] Bereavement , in which hallucinations of a deceased loved one are common [24] Severe sleep deprivation [26] [27] [28] Stress [29] Trauma [ edit ] Traumatic life events have been linked with an elevated risk in developing psychotic symptoms. [30] Childhood trauma has specifically been shown to be a predictor of adolescent and adult psychosis. [31] Approximately 65% of individuals with psychotic symptoms have experienced childhood trauma (e.g., physical or sexual abuse, physical or emotional neglect). [32] Increased individual vulnerability toward psychosis may interact with traumatic experiences promoting an onset of future psychotic symptoms, particularly during sensitive developmental periods. [31] Importantly, the relationship between traumatic life events and psychotic symptoms appears to be dose-dependent in which multiple traumatic life events accumulate, compounding symptom expression and severity. [30] [31] This suggests trauma prevention and early intervention may be an important target for decreasing the incidence of psychotic disorders and ameliorating its effects. [30] Psychiatric disorder [ edit ] From a diagnostic standpoint, organic disorders were believed to be caused by physical illness affecting the brain (that is, psychiatric disorders secondary to other conditions) while functional disorders were considered disorders of the functioning of the mind in the absence of physical disorders (that is, primary psychological or psychiatric disorders).
Some of the toxins can be present in sorghum, ragi , wheat and tomatoes. [24] [25] [26] Some research has shown that the toxins can be easily cross-contaminated between grain commodities, suggesting that manufacturing and storage of grain commodities is a critical practice. [27] Citrinin Citreoviridin Cyclopiazonic acid Cytochalasins Ergot alkaloids / ergopeptine alkaloids – ergotamine Fumonisins – Crop corn can be easily contaminated by the fungi Fusarium moniliforme , and its fumonisin B1 will cause leukoencephalomalacia (LEM) in horses, pulmonary edema syndrome (PES) in pigs, liver cancer in rats and esophageal cancer in humans. [28] [29] For human and animal health, both the FDA and the EC have regulated the content levels of toxins in food and animal feed. [30] [31] Fusaric acid Fusarochromanone Kojic acid Lolitrem alkaloids Moniliformin 3-Nitropropionic acid Nivalenol Ochratoxins – In Australia, The Limit of Reporting (LOR) level for ochratoxin A (OTA) analyses in 20th Australian Total Diet Survey was 1 µg/kg, [32] whereas the EC restricts the content of OTA to 5 µg/kg in cereal commodities, 3 µg/kg in processed products and 10 µg/kg in dried vine fruits. [33] Oosporeine Patulin – Currently, this toxin has been advisably regulated on fruit products.
Bottom , illustration of the AR protein, with primary functional domains labeled (not representative of actual 3-D structure). [2] The human androgen receptor (AR) is a protein encoded by a gene located on the proximal long arm of the X chromosome ( locus Xq11-Xq12). [9] The protein coding region consists of approximately 2,757 nucleotides (919 codons ) spanning eight exons , designated 1-8 or A-H. [4] [2] Introns vary in size between 0.7 and 26 kb . [2] Like other nuclear receptors, the AR protein consists of several functional domains : the transactivation domain (also called the transcription-regulation domain or the amino / NH2-terminal domain), the DNA-binding domain , the hinge region, and the steroid-binding domain (also called the carboxyl-terminal ligand-binding domain). [4] [10] [2] [11] The transactivation domain is encoded by exon 1, and makes up more than half of the AR protein. [2] Exons 2 and 3 encode the DNA-binding domain, while the 5' portion of exon 4 encodes the hinge region. [2] The remainder of exons 4 through 8 encodes the ligand binding domain. [2] Trinucleotide satellite lengths and AR transcriptional activity [ edit ] The AR gene contains two polymorphic trinucleotide microsatellites in exon 1. [10] The first microsatellite (nearest the 5' end) contains 8 [12] to 60 [13] [14] repetitions of the glutamine codon "CAG" and is thus known as the polyglutamine tract . [2] The second microsatellite contains 4 [15] to 31 [16] repetitions of the glycine codon "GGC" and is known as the polyglycine tract . [17] The average number of repetitions varies by ethnicity, with Caucasians exhibiting an average of 21 CAG repeats, and Blacks 18. [18] In men, disease states are associated with extremes in polyglutamine tract length; prostate cancer , [19] hepatocellular carcinoma , [20] and intellectual disability [12] are associated with too few repetitions, while spinal and bulbar muscular atrophy (SBMA) is associated with a CAG repetition length of 40 or more. [21] Some studies indicate that the length of the polyglutamine tract is inversely correlated with transcriptional activity in the AR protein, and that longer polyglutamine tracts may be associated with male infertility [22] [23] [24] and undermasculinized genitalia in men. [25] However, other studies have indicated no such correlation exists. [26] [27] [28] [29] [30] [31] A comprehensive meta-analysis of the subject published in 2007 supports the existence of the correlation, and concluded these discrepancies could be resolved when sample size and study design are taken into account. [32] Some studies suggest longer polyglycine tract lengths are also associated with genital masculinization defects in men. [33] [34] Other studies find no such association. [35] AR mutations [ edit ] As of 2010, over 400 AR mutations have been reported in the AR mutation database, and the number continues to grow. [10] Inheritance is typically maternal and follows an X-linked recessive pattern; [4] [36] individuals with a 46,XY karyotype always express the mutant gene since they have only one X chromosome , whereas 46,XX carriers are minimally affected.
Complete androgen insensitivity syndrome is a condition that affects sexual development before birth and during puberty. People with this condition are genetically male (one X and one Y chromosome) but do not respond to male hormones at all. As a result, they generally have normal female external genitalia and female breasts. However, they do not have a uterus or cervix so are unable to menstruate or conceive children. Other signs and symptoms may include undescended testes and sparse to absent pubic hair.
Interleukin 1 likely is the marker for fatigue, but increased IL-1RA is observed in the CSF and is associated with increased fatigue through cytokine -induced sickness behavior . [28] However, SS is characterized by decreased levels of IL-1ra in saliva, which could be responsible for mouth inflammation and dryness. [29] Patients with secondary SS also often exhibit signs and symptoms of their primary rheumatic disorders, such as systemic lupus erythematosus , rheumatoid arthritis , or systemic sclerosis . [ citation needed ] Genetic predisposition [ edit ] The genetic locus most significantly associated with primary SS is the major histocompatibility complex / human leukocyte antigen (MHC/HLA) region, as demonstrated by the preliminary results of the first genome-wide association study . [30] This study included data from a discovery cohort of 395 patients of European ancestry with primary SS, and 1,975 healthy control individuals , and from a replication study that comprised 1,234 cases and 4,779 healthy controls. ... "Cryoglobulinemia in primary Sjögren's syndrome: prevalence and clinical characteristics in a series of 115 patients". Semin Arthritis Rheum . 28 (3): 200–5. doi : 10.1016/S0049-0172(98)80037-1 . ... "HLA markers and clinical characteristics in Caucasians with primary Sjögren's syndrome". J. Rheumatol . 28 (7): 1554–62. PMID 11469461 . ^ Fei HM, Kang H, Scharf S, Erlich H, Peebles C, Fox R (1991).
Description Sjogren syndrome is an autoimmune disease that mainly affects the exocrine glands. It is clinically characterized by keratoconjunctivitis sicca and xerostomia (Goransson et al., 2006). See 200400 for association of Sjogren syndrome with achalasia in sisters. Clinical Features Lichtenfeld et al. (1976) noted familial occurrence. This probably represents the same sort of familial occurrence as is seen with systemic lupus erythematosus (SLE; 152700) and other autoimmune disorders.
A rare systemic autoimmune disease characterized by exocrine gland dysfunction, resulting predominately in keratoconjunctivitis sicca and xerostomia, but also affecting exocrine glands of the skin, as well as respiratory, urogenital, and digestive tract. Extraglandular manifestations include arthritis, interstitial lung disease, renal disease, and peripheral neuropathy. The disease is accompanied by a substantially increased risk to develop B-cell non-Hodgkin lymphoma, especially MALT (mucosa-associated lymphoid tissue) lymphoma.
Overview Sjogren's (SHOW-grins) syndrome is a disorder of your immune system identified by its two most common symptoms — dry eyes and a dry mouth. The condition often accompanies other immune system disorders, such as rheumatoid arthritis and lupus. In Sjogren's syndrome, the mucous membranes and moisture-secreting glands of your eyes and mouth are usually affected first — resulting in decreased tears and saliva. Although you can develop Sjogren's syndrome at any age, most people are older than 40 at the time of diagnosis. The condition is much more common in women. Treatment focuses on relieving symptoms.
Sjögren syndrome is a disorder whose main features are dry eyes and a dry mouth. The condition typically develops gradually beginning in middle adulthood, but it can occur at any age. Sjögren syndrome is classified as an autoimmune disorder, one of a large group of conditions that occur when the immune system attacks the body's own tissues and organs. In Sjögren syndrome, the immune system primarily attacks the glands that produce tears (the lacrimal glands) and saliva (the salivary glands), impairing the glands' ability to secrete these fluids. Dry eyes may lead to itching, burning, a feeling of sand in the eyes, blurry vision, or intolerance of bright or fluorescent lighting.