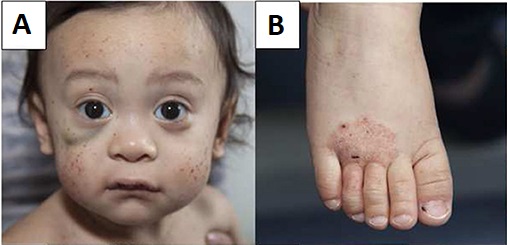
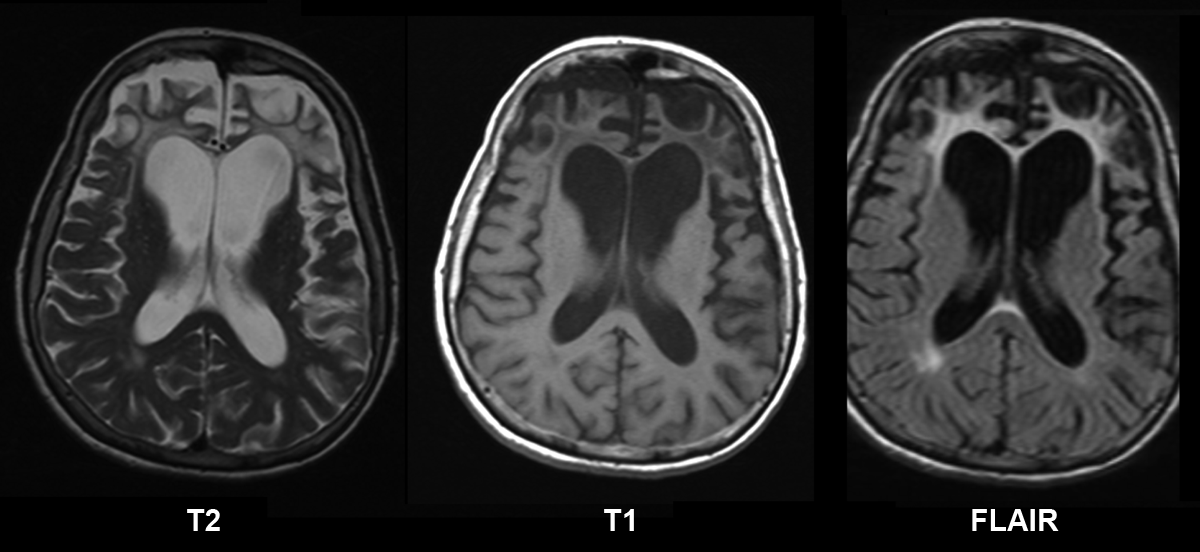
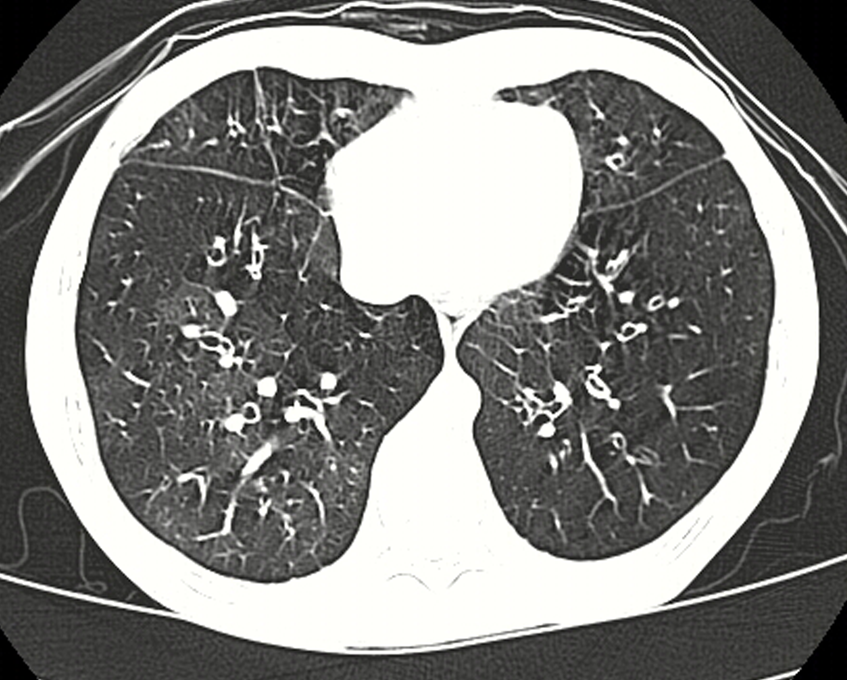
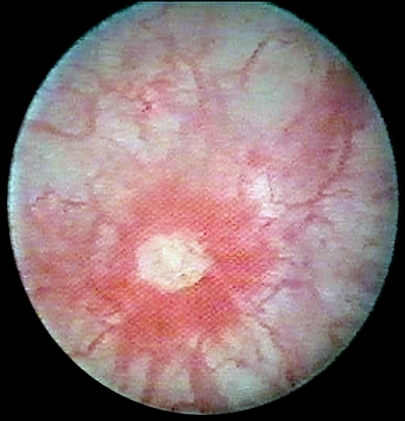
The reported age of cataract extraction ranges from 28 to 74 years [Day et al 2003]. With aging, an increase in macular thickness based on epiretinal membranes can lead to visual impairment.
Myotonic dystrophy type 2, one of the two types of myotonic dystrophy , is an inherited muscular dystrophy that affects the muscles and other body systems (e.g., heart, eyes, and pancreas). It is characterized by prolonged muscle tensing ( myotonia ) as well as muscle weakness, pain, and stiffness. Signs and symptoms usually develop during a person's twenties or thirties. Muscles in the neck, fingers, elbows, and hips are typically affected; facial and ankle muscles are less commonly involved. The severity of myotonic dystrophy type 2 varies widely among affected people, even among family members.
A rare myotonic dystrophy of juvenile or adult-onset characterized by mild and fluctuating myotonia, muscle weakness, and rarely cardiac conduction disorders. Epidemiology Prevalence estimates of around 1/100,000 in Germany and 1/600,000 in the United Kingdom have been suggested. Most cases have been reported in ethnic Europeans (mainly in Eastern Europe with a founder effect). Clinical description The clinical presentation is marked by significant variability as is the age of onset (juvenile to late adulthood). The primary manifestations include muscle weakness (with early involvement of neck flexors or finger flexors, later involving hip girdle muscles), myotonia, and possibly severe fluctuating or episodic muscle pain, and stiffness.
A number sign (#) is used with this entry because myotonic dystrophy-2 (DM2/PROMM) is caused by heterozygous expansion of a CCTG repeat in intron 1 of the zinc finger protein-9 gene (ZNF9; 116955). Normal ZNF9 alleles have up to 30 repeats; pathogenic alleles contain from 75 to 11,000 repeats (Todd and Paulson, 2010). Description Myotonic dystrophy (DM) is a multisystem disorder and the most common form of muscular dystrophy in adults. Individuals with DM2 have muscle pain and stiffness, progressive muscle weakness, myotonia, male hypogonadism, cardiac arrhythmias, diabetes, and early cataracts. Other features may include cognitive dysfunction, hypersomnia, tremor, and hearing loss (summary by Heatwole et al., 2011).
Gene therapy For severely affected males without an HLA-matched donor, studies of correcting Wiskott–Aldrich syndrome with gene therapy using a lentivirus are underway. [28] [29] Proof-of-principle for successful hematopoietic stem cell gene therapy has been provided for patients with Wiskott–Aldrich syndrome. [30] In July 2013 the Italian San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET) reported that three children with Wiskott–Aldrich syndrome showed significant improvement (improved platelet counts, immune functiona, and clinical symptoms) 20–30 months after being treated with a genetically modified lentivirus. [31] In April 2015 results from a follow-up British and French trial six out of seven individuals showed improvement of immune function and clinical symptoms an average of 27 months after treatment with gene therapy. [32] [33] [34] Importantly, neither study showed evidence of leukemic proliferation following treatment, a complication of early attempts at gene therapy using a retroviral vector. [35] It is unknown why these gene therapies did not restore normal platelet numbers, but gene therapy treatment was still associated with transfusion-independence and a significant reduction in bleeding events. [31] [32] Prognosis [ edit ] Outcomes from Wiskott–Aldrich syndrome are variable and depend on how severely an individual is affected (the WAS score may be used to assess disease severity).
A number sign (#) is used with this entry because of evidence that Wiskott-Aldrich syndrome-2 (WAS2) is caused by homozygous mutation in the WIPF1 gene (602357) on chromosome 2q31. One such patient has been reported. For a discussion of genetic heterogeneity of Wiskott-Aldrich syndrome, see WAS (301000). Clinical Features Conley et al. (1992) raised the possibility of the existence of an autosomal recessive form of Wiskott Aldrich syndrome. They observed an 8-year-old girl with a disorder phenotypically identical to the disorder in males with an X-linked mutation (WAS; 301000). Cytogenetic studies showed no structural abnormalities of the X chromosome and X-chromosome inactivation analysis showed that both X chromosomes could function as the active X.
A primary immunodeficiency disease characterized by microthrombocytopenia, eczema, infections and an increased risk for autoimmune manifestations and malignancies. Epidemiology The incidence of WAS has been estimated at less than 1 in 100,000 live births. The disease almost exclusively affects males. Clinical description WAS usually manifests in infancy but onset may also occur during the neonatal period. In most cases the first clinical features are hemorrhagic manifestations with petechiae, bruising, purpura, epistaxis, oral bleeding, bloody diarrhea and intracranial bleeding. Acute or chronic eczema is the second characteristic finding of WAS. Due to combined immunodeficiency, most patients also have airway, gut or skin infections caused by regular or opportunistic germs.
Wiskott-Aldrich syndrome is characterized by abnormal immune system function (immune deficiency), eczema (an inflammatory skin disorder characterized by abnormal patches of red, irritated skin), and a reduced ability to form blood clots . This condition primarily affects males. Individuals with Wiskott-Aldrich syndrome have microthrombocytopenia, which is a decrease in the number and size of blood cells involved in clotting (platelets). This platelet abnormality, which is typically present from birth, can lead to easy bruising, bloody diarrhea, or episodes of prolonged bleeding following nose bleeds or minor trauma. Microthrombocytopenia can also lead to small areas of bleeding just under the surface of the skin, resulting in purplish spots called purpura, or variably sized rashes made up of tiny red spots called petechiae. In some cases, particularly if a bleeding episode occurs within the brain, prolonged bleeding can be life-threatening.
Wiskott Aldrich syndrome (WAS) is a disease with immunological deficiency and reduced ability to form blood clots. Signs and symptoms include easy bruising or bleeding due to a decrease in the number and size of platelets; susceptibility to infections and to immune and inflammatory disorders; and an increased risk for some cancers (such as lymphoma ). Also, a skin condition known as eczema is common in people with WAS. Wiskott Aldrich syndrome is caused by mutations in the WAS gene and is inherited in an X-linked manner. It primarily affects males. Treatment may depend on severity and symptoms in each person, but hematopoietic cell transplantation is the only known cure. Hematopoietic cells are the blood-forming stem cells that can be found mainly in the sponge-like material found inside bones (bone marrow), but also in the bloodstream (peripheral blood stem cells (PBSCs), and in the umbilical cord.
A number sign (#) is used with this entry because Wiskott-Aldrich syndrome (WAS) is caused by mutation in the WAS gene (300392) on chromosome Xp11. Description Wiskott-Aldrich syndrome (WAS) is an X-linked recessive immunodeficiency characterized by thrombocytopenia, eczema, and recurrent infections (Lemahieu et al., 1999). Genetic Heterogeneity of Wiskott-Aldrich Syndrome See Wiskott-Aldrich syndrome-2 (WAS2; 614493), caused by mutation in the WIPF1 gene (602357). Also see 600903 for a possible autosomal dominant form of the disorder. Clinical Features The manifestations of Wiskott-Aldrich syndrome are eczema, thrombocytopenia, proneness to infection, and bloody diarrhea.
Neri et al. (1995) raised the possibility of an autosomal dominant form of Wiskott-Aldrich syndrome on the basis of a 3-generation family in which several members presented clinical and laboratory findings of WAS (301000), including decreased CD43 expression on T lymphocytes. The gene for CD43, or sialophorin (SPN; 182160), is located on 16p11.2. However, no alteration of CD43 was found: Southern blot analysis failed to detect gross abnormalities of the CD43 gene and genotype analysis showed that the affected family members did not share a common CD43 allele. INHERITANCE - Autosomal dominant - Autosomal recessive HEAD & NECK Head - Sinusitis Ears - Otitis media Nose - Epistaxis Mouth - Oral bleeding CARDIOVASCULAR Vascular - Small and large vessel vasculitis RESPIRATORY Airways - Upper respiratory tract infections - Lower respiratory tract infections Lung - Pneumonia ABDOMEN Gastrointestinal - Diarrhea - Inflammatory bowel disease GENITOURINARY Kidneys - Nephropathy SKIN, NAILS, & HAIR Skin - Eczema NEUROLOGIC Central Nervous System - Meningitis HEMATOLOGY - Thrombocytopenia - Small platelet size - Hemolytic anemia - Iron deficiency anemia - CD43 (sialophorin) defectively expressed on surface of blood cells IMMUNOLOGY - Moderately depressed antibody response to polysaccharide antigens - Lymphopenia - Abnormal delayed hypersensitivity skin test - Absent microvilli on the surface of peripheral blood lymphocytes - Decreased CD3+ cells subset - Decreased CD4+ cells subset - Decreased CD8+ cells subset LABORATORY ABNORMALITIES - Prolonged bleeding time - Normal IgG levels - Increased IgA levels - Increased IgE levels - Reduced IgM levels - Raised erythrocyte sedimentation rate - Raised C-reactive protein MISCELLANEOUS - Normal sialophorin gene ▲ Close
Hb Bart syndrome should be suspected in the following: An at-risk fetus with increased nuchal thickness, thickened placenta, increased cerebral media artery velocity, and increased cardiothoracic ratio on ultrasonography examination at 13-14 weeks' gestation A fetus with generalized edema, ascites, and pleural and pericardial effusions on ultrasonography examination at 22-28 weeks' gestation HbH disease should be suspected in an infant or child with the following clinical or newborn screening findings: Clinical findings Mild jaundice Hepatosplenomegaly Mild thalassemia-like bone changes (e.g., hypertrophy of the maxilla, bossing of the skull, and prominence of the malar eminences) Newborn screening findings .
Alpha thalassemia is a blood disorder that reduces the production of hemoglobin . Hemoglobin is the protein in red blood cells that carries oxygen to cells throughout the body. In people with the characteristic features of alpha thalassemia, a reduction in the amount of hemoglobin prevents enough oxygen from reaching the body's tissues. Affected individuals also have a shortage of red blood cells (anemia ), which can cause pale skin, weakness, fatigue, and more serious complications. Two types of alpha thalassemia can cause health problems. The more severe type is known as hemoglobin Bart hydrops fetalis syndrome, which is also called Hb Bart syndrome or alpha thalassemia major.
Hemoglobin H (HbH) disease is a moderate to severe form of alpha-thalassemia (see this term) characterized by pronounced microcytic hypochromic hemolytic anemia. Epidemiology HbH disease is predominantly seen in Southeast Asia, the Middle East and the Mediterranean. Exact prevalence is not known but has been reported to be about 1/1,000,000 for severe forms of alpha-thalassemia in Northern Europe and North America. Clinical description Clinical features are highly variable and generally develop in the first years of life but may not develop until adulthood in some patients. Initial signs may be noticed only during routine hematologic analyses.
An inherited hemoglobinopathy characterized by impaired synthesis of alpha-globin chains leading to a variable clinical picture depending on the number of affected alleles. Epidemiology Like other globin gene disorders, alpha-thalassemia is highly prevalent in all tropical and subtropical regions (around 1/10,000), particularly in the African equatorial belt. Intermediate and severe forms of alpha-thalassemia are very rare in North America and Northern Europe (about 1/1,000,000), and are predominantly seen in immigrant populations from South-East Asia or Mediterranean countries. Clinical description The disease can be classified into clinical subtypes of increasing severity: silent alpha thalassemia, alpha thalassemia trait (or alpha thalassemia minor), hemoglobin H disease (HbH), and Hb Bart's hydrops fetalis. A rare form called alpha-thalassemia-intellectual deficit syndrome linked to chromosome 16 (16p13.3) has also been identified (see these terms).
A number sign (#) is used with this entry because of evidence that alpha-thalassemia is caused by mutations in the alpha-globin genes (HBA1, 141800; HBA2, 141850). Sequences 30 to 50 kb upstream from the alpha-globin gene cluster, referred to as the locus control region alpha (LCRA; 152422), have been found to be deleted in cases of alpha-thalassemia with structurally intact alpha-globin genes. The molecular and clinical aspects of the severe alpha-thalassemia syndromes were reviewed by Higgs (1993) and Chui and Waye (1998). Weatherall (2001) reviewed phenotype-genotype relationships in monogenic diseases based on studies of the thalassemias. The remarkable phenotypic diversity of the beta-thalassemias reflects the heterogeneity of mutations at the HBB locus, the action of many secondary and tertiary modifiers, and a wide range of environmental factors.
Alpha-thalassemia is a blood disorder that reduces the body's production of hemoglobin . Affected people have anemia, which can cause pale skin, weakness, fatigue, and more serious complications. Two types of alpha-thalassemia can cause health problems: the more severe type is known as Hb Bart syndrome; the milder form is called HbH disease. Hb Bart syndrome may be characterized by hydrops fetalis ; severe anemia; hepatosplenomegaly; heart defects; and abnormalities of the urinary system or genitalia. Most babies with this condition are stillborn or die soon after birth.
Meta-analyses based on imaging methods have shown that frontotemporal dementia mainly affects a frontomedial network discussed in the context of social cognition or ' theory of mind '. [27] This is entirely in keeping with the notion that on the basis of cognitive neuropsychological evidence, the ventromedial prefrontal cortex is a major locus of dysfunction early on in the course of the behavioural variant of frontotemporal degeneration. [28] The language subtypes of frontotemporal lobar degeneration (semantic dementia and progressive nonfluent aphasia) can be regionally dissociated by imaging approaches in vivo . [29] The confusion between Alzheimer's and FTD is justifiable due to the similarities between their initial symptoms.
Frontotemporal dementia with parkinsonism-17 (FTDP-17) is a brain disorder. It is part of a group of conditions, called frontotemporal dementia or frontotemporal degeneration, that are characterized by a loss of nerve cells (neurons ) in areas of the brain called the frontal and temporal lobes. Over time, a loss of these cells can affect personality, behavior, language, and movement. The signs and symptoms of FTDP-17 usually become noticeable in a person's forties or fifties. Most affected people survive 5 to 10 years after the appearance of symptoms, although a few have survived for two decades or more.
Frontotemporal dementias (FTDs) are a group of neurodegenerative disorders associated with shrinking of the frontal and temporal anterior lobes of the brain. Symptoms include marked changes in social behavior and personality, and/or problems with language. People with behavior changes may have disinhibition (with socially inappropriate behavior), apathy and loss of empathy, hyperorality (eating excessive amounts of food or attempting to consume inedible things), agitation, compulsive behavior, and various other changes. Examples of problems with language include difficulty speaking or understanding speech. Some people with FTD also develop a motor syndrome such as parkinsonism or motor neuron disease (which may be associated with various additional symptoms).
Overview Frontotemporal dementia is an umbrella term for a group of brain disorders that primarily affect the frontal and temporal lobes of the brain. These areas of the brain are generally associated with personality, behavior and language. In frontotemporal dementia, portions of these lobes shrink (atrophy). Signs and symptoms vary, depending on which part of the brain is affected. Some people with frontotemporal dementia have dramatic changes in their personalities and become socially inappropriate, impulsive or emotionally indifferent, while others lose the ability to use language properly.
Frontotemporal dementia (FTD) comprises a group of neurodegenerative disorders, characterized by progressive changes in behavior, executive dysfunction and language impairment, as a result of degeneration of the medial prefrontal and frontoinsular cortices. Four clinical subtypes have been identified: semantic dementia, progressive non-fluent aphasia, behavioral variant FTD and right temporal lobar atrophy (see these terms).
Summary Clinical characteristics. The spectrum of GRN frontotemporal dementia ( GRN -FTD) includes the behavioral variant (bvFTD), primary progressive aphasia (PPA; further subcategorized as progressive non-fluent aphasia [PNFA] and semantic dementia [SD]), and movement disorders with extrapyramidal features such as parkinsonism and corticobasal syndrome (CBS). A broad range of clinical features both within and between families is observed. The age of onset ranges from 35 to 87 years. Behavioral disturbances are the most common early feature, followed by progressive aphasia. Impairment in executive function manifests as loss of judgment and insight. In early stages, PPA often manifests as deficits in naming, word finding, or word comprehension.
A number sign (#) is used with this entry because of evidence that frontotemporal dementia mapping to chromosome 3 is caused by heterozygous mutation in the CHMP2B gene (609512) on chromosome 3p11. Mutation in the CHMP2B gene can also cause a form of amyotrophic lateral sclerosis (ALS17; 614696). Description A substantial minority of degenerative dementias, perhaps 10%, lack the distinctive pathologic features that allow subclassification as Alzheimer disease (see 104300) or other forms of dementia. In perhaps half of these cases of nonspecific dementia, there is a positive family history of dementia, with an apparent autosomal dominant mode of inheritance. See also frontotemporal lobe dementia (FLDEM; 600274), which maps to chromosome 17 and is caused by mutation in the microtubule-associated protein tau gene (MAPT; 157140).
If AQP4-IgG is detected, then 1 core clinical criterion, along with the ruling out of alternative diagnoses , is sufficient for NMOSD diagnosis. [24] If AQP4-IgG is undetected or its status unknown, 2 core clinical criteria, each with supportive MRI findings, along with the ruling out of alternative diagnoses, are needed for NMOSD diagnosis. [25] NMOSD diagnostic criteria [24] Core criteria Additional MRI findings for absent/unknown AQP4-IgG Optic neuritis Either 1) brain MRI showing normal findings or only nonspecific white matter lesions, or 2) optic nerve MRI showing T2-hyperintesity, or T1 enhancing lesion, greater than 1/2 optic nerve length or involving optic chiasm Acute myelitis intramedullary lesion > 3 contiguous segments, or spinal atrophy ≥ 3 contiguous segments Area Postrema Syndrome (prolonged episodes of hiccuping or vomiting / nausea ) dorsal medulla/area postrema lesions Acute brainstem syndrome periependymal brainstem lesions Symptomatic narcolepsy /acute diencephalic clinical syndrome with a MRI showing diencephalon lesion (s) None additional Symptomatic cerebral syndrome with NSMOD-typical brain lesion (s) None additional Rarely, it has been reported that some courses of anti-NMDAR are consistent with NMO. [26] Preliminary reports suggest that other autoantibodies may play a role in rare cases of NMO. [27] [28] NMOSD with MOG-IgG is considered a manifestation of anti-MOG associated encephalomyelitis . [29] Spectrum constituents [ edit ] After the development of the NMO- IgG test, the spectrum of disorders comprising NMO was expanded. ... Food and Drug Administration (FDA) (Press release). 27 June 2019 . Retrieved 28 June 2019 . ^ "FDA Approves New Therapy for Rare Disease Affecting Optic Nerve, Spinal Cord" . ... Anthony, Jacqueline Palace, Classifying the antibody-negative NMO syndromes, Clinical, imaging, and metabolomic modeling, October 28, 2019, DOI: https://doi.org/10.1212/NXI.0000000000000626 External links [ edit ] Classification D ICD - 10 : G36.0 ICD - 9-CM : 341.0 MeSH : D009471 DiseasesDB : 29470 External resources Orphanet : 71211 v t e Multiple sclerosis and other demyelinating diseases of the central nervous system Signs and symptoms Ataxia Depression Diplopia Dysarthria Dysphagia Fatigue Incontinence Nystagmus Optic neuritis Pain Uhthoff's phenomenon Investigations and diagnosis Multiple sclerosis diagnosis McDonald criteria Poser criteria Clinical Clinically isolated syndrome Expanded Disability Status Scale Serological and CSF Oligoclonal bands Radiological Radiologically isolated syndrome Lesional demyelinations of the central nervous system Dawson's fingers Approved [ by whom?
Neuromyelitis optica spectrum disorders (NMOSD) affect the spinal cord and optic nerves (nerves that carry visual messages to and from the brain). Symptoms include pain, weakness, bowel and bladder problems, and temporary vision loss. NMOSD usually occurs in adulthood, but symptoms may start at any age. Some people have a single attack of symptoms lasting months, but in most people the symptoms come and go over time. People with NMOSD may develop permanent muscle weakness and vision loss.
Neuromyelitis optica is an autoimmune disorder that affects the nerves of the eyes and the central nervous system, which includes the brain and spinal cord. Autoimmune disorders occur when the immune system malfunctions and attacks the body's own tissues and organs. In neuromyelitis optica, the autoimmune attack causes inflammation of the nerves, and the resulting damage leads to the signs and symptoms of the condition. Neuromyelitis optica is characterized by optic neuritis, which is inflammation of the nerve that carries information from the eye to the brain (optic nerve ). Optic neuritis causes eye pain and vision loss, which can occur in one or both eyes.
Overview Neuromyelitis optica (NMO) is a central nervous system disorder that causes inflammation in nerves of the eye and the spinal cord. NMO is also called neuromyelitis optica spectrum disorder (NMOSD) and Devic disease. It occurs when the body's immune system reacts against its own cells. This happens mainly in the optic nerves that connect the retina of the eye with the brain and in the spinal cord. But it sometimes occurs in the brain. The disorder may appear after an infection.
Neuromyelitis optica (NMO) and NMO spectrum disorders are inflammatory demyelinating diseases of the central nervous system characterized mainly by attacks of uni- or bilateral optic neuritis (ON) and acute myelitis. Epidemiology NMO has a worldwide distribution and estimated prevalence of 1-2/100,000. Clinical description Patients present with acute, often severe, attacks of blindness and paraparesis or quadriparesis, accompanied by sensory and sphincter impairments. Most patients have relapsing attacks (separated by months or years with partial recovery), with usually sequential index episodes of ON and myelitis. A relapsing course is more frequent in women, and nearly 90% of patients are female (typically late middle-aged).
Among 148 Ashkenazi Jews carrying the Tay-Sachs gene, Grebner and Tomczak (1991) found that 108 had the insertion mutation (606869.0001), 26 had the splice junction mutation (606869.0002), 5 had the adult mutation (606869.0008), and 9 had none of the 3. Among 28 non-Jewish carriers tested, most of whom were obligate carriers, 4 had the insertion mutation, 1 had the adult mutation, and the remaining 23 had none of the 3.
Tay -Sachs disease is a rare, inherited neurodegenerative disease . People with Tay-Sachs disease do not have enough of an enzyme called beta-hexosaminidase A. The less enzyme a person has, the more severe the disease and the earlier that symptoms appear. There are 3 forms of Tay-Sachs disease, distinguished by the general age of onset: Infantile - the most common severe form, with symptoms appearing in the first few months of life. Symptoms include a loss of skills learned (regression), seizures, and loss of muscle and mental functions. Children with this form do not survive past early childhood. Juvenile - a form with a range of severity, with symptoms appearing any time during childhood (but usually between ages 2 and 5).
GM2 gangliosidoses Specialty Endocrinology The GM2 gangliosidoses are a group of three related genetic disorders that result from a deficiency of the enzyme beta-hexosaminidase . This enzyme catalyzes the biodegradation of fatty acid derivatives known as gangliosides . [1] The diseases are better known by their individual names: Tay–Sachs disease , AB variant , and Sandhoff disease . Beta-hexosaminidase is a vital hydrolytic enzyme, found in the lysosomes , that breaks down lipids. When beta-hexosaminidase is no longer functioning properly, the lipids accumulate in the nervous tissue of the brain and cause problems. Gangliosides are made and biodegraded rapidly in early life as the brain develops.
Overview Tay-Sachs disease is a rare genetic disorder passed from parents to child. It's caused by the absence of an enzyme that helps break down fatty substances. These fatty substances, called gangliosides, build up to toxic levels in the brain and spinal cord and affect the function of the nerve cells. In the most common and severe form of Tay-Sachs disease, signs and symptoms start to show up at about 3 to 6 months of age. As the disease progresses, development slows and muscles begin to weaken.
Tay-Sachs disease is a rare inherited disorder that progressively destroys nerve cells (neurons ) in the brain and spinal cord. The most common form of Tay-Sachs disease becomes apparent in infancy. Infants with this disorder typically appear normal until the age of 3 to 6 months, when their development slows and muscles used for movement weaken. Affected infants lose motor skills such as turning over, sitting, and crawling. They also develop an exaggerated startle reaction to loud noises. As the disease progresses, children with Tay-Sachs disease experience seizures, vision and hearing loss, intellectual disability, and paralysis.
A rare disorder characterized by accumulation of G2 gangliosides due to hexosaminidase A deficiency. Epidemiology The prevalence of the disease is 1 case per 320 000 live births. Clinical description Three variants have been described according to age of onset. The infantile form (type 1) begins between 3 and 6 months of age. The earliest sign is an incessant startle response to noise. Psychomotor retardation appears after the age of 8 months with hypotonia, amaurosis, and megalencephaly.
Due to this event, obliterative bronchiolitis began to be referred to in the popular media as "popcorn lung" or "popcorn workers lung". [26] [27] [28] [29] It is also referred to as "flavorings-related lung disease". [30] Post-infectious [ edit ] High resolution CT scan of a child with post-infectious obliterative bronchiolitis showing glass pattern with air trapping and bronchial thickening Typically found in young children and is the most common cause at this age. [31] Generally occurs after a viral infection of adenovirus (types 3, 7, and 21), measles (rubeola), mycoplasma, CMV, influenza, and parainfluenza. [4] [6] Swyer-James syndrome is a rare complication of obliterative bronchiolitis caused by measles or adenovirus. [32] Post-infectious obliterative bronchiolitis is most common in the southern hemisphere particularly in countries such as Brazil, Argentina, Australia, Chile and New Zealand. [33] There was a large prevalence of the disease in these areas during the 1990s and early 2000s.
Bronchiolitis obliterans is an inflammatory condition that affects the lung's tiniest airways, the bronchioles. In affected people, the bronchioles may become damaged and inflamed leading to extensive scarring that blocks the airways. Signs and symptoms of the condition include a dry cough; shortness of breath; and/or fatigue and wheezing in the absence of a cold or asthma. Many different chemicals (such as nitrogen oxides, ammonia, welding fumes or food flavoring fumes) and respiratory infections can cause lung injury that leads to bronchiolitis obliterans. It can also be associated with rheumatoid arthritis and graft-versus-host disease following a lung or hematopoietic cell transplantation .
Bronchiolitis obliterans syndrome (BOS) is a lung disorder that is mainly associated with chronic allograft dysfunction after lung transplantation and that is characterized by inflammation and fibrosis of bronchiolar walls that reduce the diameter of the bronchioles and result in progressive and irreversible airflow obstruction.
MRI is the diagnostic modality of choice for imaging prior to biopsy and pathologic diagnosis, with the primary role being the determination of anatomic boundaries. [ citation needed ] Staging [ edit ] The staging for epithelioid sarcoma takes into account size and location of the primary tumor, lymph node involvement, presence and location of metastasis, and histologic grade (a measure of disease aggressiveness) [26] Treatment [ edit ] Surgical resection of the tumor with wide margins remains the preferred method of treatment, [27] and has shown the most success against the disease. [27] [28] [29] Recently, limb-sparing surgery has been explored with moderate success. [30] In cases of advanced, recurrent, or metastasized disease, or if the tumor is inoperable, chemotherapy and radiation are the standard of care, [31] although the overall success rates with these remains low. [32] In January 2020, The U.S. ... "Targeting nucleocytoplasmic transport in cancer therapy" . Oncotarget . 5 (1): 11–28. doi : 10.18632/oncotarget.1457 .
Epithelioid sarcoma (ES) is a rare cancerous tumor that most often occurs in the soft tissue of the fingers, hands and forearms of young adults. It can also occur elsewhere in the body. ES usually begins as a painless, firm growth or bump that may be accompanied by an open wound ( ulceration ) in the skin covering the growth. This type of tumor often comes back after treatment or spreads to other parts the body (metastasis). The cause of epithelioid sarcoma is unknown. It is diagnosed by a clinical examination and by testing a small sample of the tumor (biopsy) in a laboratory. Epithelioid sarcoma is treated with surgery to remove all the cancer cells ( wide local excision ) and sometimes with radiation therapy.
Epithelioid sarcoma is a rare, soft tissue tumor characterized by high incidence of local recurrence, regional lymph node involvement and distant metastases. It commonly affects the soft tissue under the skin of a finger, hand, forearm, lower leg or foot, less often other areas of the body.
The body attacks itself, which is the basis of autoimmune disorders. [28] Additionally, IC may be triggered by an unknown toxin or stimulus which causes nerves in the bladder wall to fire uncontrollably.
Overview Interstitial cystitis (in-tur-STISH-ul sis-TIE-tis) is a chronic condition causing bladder pressure, bladder pain and sometimes pelvic pain. The pain ranges from mild discomfort to severe pain. The condition is a part of a spectrum of diseases known as painful bladder syndrome. Your bladder is a hollow, muscular organ that stores urine. The bladder expands until it's full and then signals your brain that it's time to urinate, communicating through the pelvic nerves. This creates the urge to urinate for most people. Interstitial cystitis Your bladder, kidneys, ureters and urethra make up your urinary system. When you have interstitial cystitis, the walls of your bladder become irritated and inflamed (shown right), compared with those of a normal bladder (shown top).
A rare non-infectious, chronic and most often progressive disease of the urinary bladder. It is characterized by varying combinations and extent of pain, urinary frequency (pollakisuria), nocturia and urgencyInterstitial cystitis (IC) has a broad intersection with Bladder Pain Syndrome (BPS) and Overactive Bladder (OAB). Epidemiology Prevalence of IC/BPS is approximately 1/200-2,000- for females and 1/2,450-12,500 for males. The prevalence of IC alone is less than 1/2,000. The estimated number of unreported cases might be much higher. Women are more affected than men (ratio 9:1). Clinical description IC is characterized by a permanent or intermittent unpleasant sensation (pain, pressure, bladder spasms) in the pelvic (or vulvar, suprapubic, pubic, vaginal, perineal, scrotal or urethral) region of more than 6 month duration, often with worsening during bladder filling.
TSC-LAM occurs in women who have germline mutations in either the TSC1 or the TSC2 gene. [20] Sporadic LAM is primarily associated with somatic TSC2 gene mutations. [21] [22] Germline and somatic mutations in LAM include many types of mutations spread across the genes, with no clear “hot spots,” including missense changes, in-frame deletions and nonsense mutations. [20] [21] [22] Because of the large size of the genes (together they have more than 60 exons) and because mutations can be located virtually anywhere within the genes, mutation detection is often challenging. [ citation needed ] On a cellular basis, LAM cells carry bi-allelic inactivation of the TSC2 genes, consistent with the “two-hit” tumor suppressor gene model. [23] [24] The second hit event in LAM cells is often loss of the chromosomal region containing the wild-type copy of the TSC2 gene; this is referred to as loss of heterozygosity or LOH. [25] LOH can be detected in microdissected LAM cells, [21] [26] in angiomyolipomas and lymph nodes from women with LAM, [27] and in circulating LAM cells (cells in blood and urine). [28] [29] Angiomyolipomas and pulmonary LAM cells from women with the sporadic form of LAM carry identical mutations in TSC2. [21] This, together with the fact that recurrent LAM after lung transplantation carries the same TSC2 mutations as the original LAM, [30] has led to the "benign metastasis" hypothesis that LAM cells can migrate or metastasize from one site to another. [17] [18] Pathophysiology [ edit ] A variable percentage of cells within the LAM lesion contain mutational inactivation of the Tuberous Sclerosis Complex (TSC1 or TSC2) tumor suppressor genes. [21] [27] [31] TSC1 mutations cause a less severe clinical phenotype than TSC2 mutations. [32] The discovery of TSC1/2 gene function as negative regulator of the mammalian target of rapamycin complex 1 (mTORC1) [33] [34] led to successful use of rapamycin analog sirolimus in clinical trials [35] [36] and FDA approval of sirolimus for treatment of LAM. ... "Mutation analysis of the TSC1 and TSC2 genes in Japanese patients with pulmonary lymphangioleiomyomatosis" . J Hum Genet . 47 (1): 20–28. doi : 10.1007/s10038-002-8651-8 .
A number sign (#) is used with this entry because lymphangioleiomyomatosis (LAM) can occur in association with tuberous sclerosis complex (TSC; 191100) due to mutations in the TSC1 (605284) or TSC2 (191092) genes. Sporadic LAM typically results from 2 somatic mutations in the TSC2 gene, although a fraction of sporadic LAM is caused by germline mutations in the TSC1 gene. Clinical Features Pulmonary lymphangiomyomatosis, also known as pulmonary lymphangioleiomyomatosis (Urban et al., 1999), is a rare disease that occurs almost exclusively in women. It was first described by Van Stossel (1937). The average age at onset of symptoms, which include shortness of breath (67%), lung collapse (25%), coughing (12%), and chest pain (10%), is 33 years (Taylor et al., 1990; Johnson et al., 1993). Chest x-rays typically show a diffuse interstitial infiltrate, with no predominance in any 1 lung zone.
Lymphangioleiomyomatosis (LAM) is a condition that affects the lungs, the kidneys , and the lymphatic system . The lymphatic system consists of a network of vessels that transport lymph fluid and immune cells throughout the body. Lymph fluid helps exchange immune cells, proteins, and other substances between the blood and tissues. LAM is found almost exclusively in women. It often occurs as a feature of an inherited syndrome called tuberous sclerosis complex. When LAM occurs alone it is called isolated or sporadic LAM. Signs and symptoms of LAM most often appear during a woman's thirties.
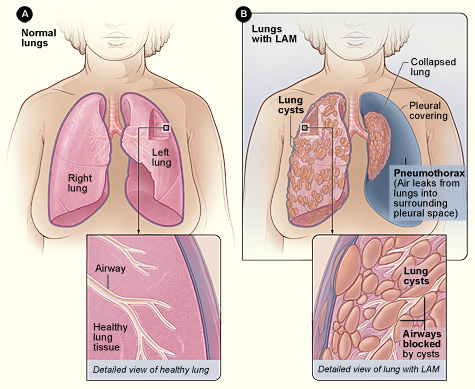
Lymphangioleiomyomatosis (LAM) is a multiple cystic lung disease characterized by progressive cystic destruction of the lung and lymphatic abnormalities, frequently associated with renal angiomyolipomas (AMLs). LAM occurs either sporadically or as a manifestation of tuberous sclerosis complex (TSC). Epidemiology Sporadic LAM affects around 1/500,000-1/125,000 adult women in Europe. TSC is present in 1 in 6,000 births. Pulmonary LAM is present in up to 30-40% of adult TSC cases. LAM affects almost exclusively females. Clinical description Defining manifestations of the disease are respiratory and include progressive dyspnea, pneumothorax and chylothorax.
Lymphangioleiomyomatosis (lim-FAN-je-o-LI-o-MI-o-ma-TO-sis), or LAM, is a rare cystic lung disease that mostly affects women in their mid-forties. In LAM, an unusual type of cell begins to grow out of control throughout the body, including in the lungs, lymph nodes and vessels, and kidneys. Over time, these LAM cells form cysts and clusters of cells, which grow throughout the lungs and slowly block the airways. They also destroy the normal lung tissue and replace it with cysts. As a result, air cannot move freely in and out of the lungs, and the lungs cannot supply enough oxygen to the body’s other organs. Some people also develop growths called angiomyolipomas (AMLs) in the kidneys.
Another drug in development is targeting VWF and its binding sites, thereby reducing VWF-platelet interaction, especially on ULVWF during a TTP episode. [26] Among several (multi-)national data bases a worldwide project has been launched to diagnose USS patients and collect information about them to gain new insights into this rare disease with the goal to optimize patient care. [27] Epidemiology [ edit ] The incidence of acute TTP in adults is around 1.7–4.5 per million and year. [28] [29] These cases are nearly all due to the autoimmune form of TTP, where autoantibodies inhibit ADAMTS13 activity. [1] [9] The prevalence of USS has not yet been determined, but is assumed to constitute less than 5% of all acute TTP cases.
Congenital thrombotic thrombocytopenic purpura (congenital TTP) is a blood disorder in which blood clots form in the small blood vessels throughout the body. Signs and symptoms typically develop in infancy or early childhood, but in some cases they do not develop until adulthood, particularly during pregnancy in women or after an infection or vaccination. Signs and symptoms generally are due to hemolytic anemia, low platelets (thrombocytopenia), and neurologic dysfunction. Symptoms of anemia can include fatigue, paleness, jaundice, shortness of breath, and a rapid heart rate. Widespread thrombosis (abnormal clotting) can lead to problems with the nervous system (such as personality changes, headaches, confusion, and seizures), abnormal kidney function, heart problems, and gastrointestinal problems.
Thrombotic thrombocytopenic purpura is a rare disorder that causes blood clots (thrombi) to form in small blood vessels throughout the body. These clots can cause serious medical problems if they block vessels and restrict blood flow to organs such as the brain, kidneys, and heart. Complications resulting from these clots can include neurological problems (such as personality changes, headaches, confusion, and slurred speech), fever, abnormal kidney function, abdominal pain, and heart problems. Blood clots normally form to stop blood loss at the sites of blood vessel injury. In people with thrombotic thrombocytopenic purpura, clots develop even in the absence of apparent injury.
A hereditary form of thrombotic thrombocytopenic purpura (TTP) characterized by profound peripheral thrombocytopenia, microangiopathic hemolytic anemia (MAHA) and single or multiple organ failure of variable severity. Epidemiology Congenital TTP is much less common than the immune-mediated form of the disease (immune-mediated TTP), accounting for up to only 5% of all TTP cases. Up until 2017, 123 cases had been reported by the International Hereditary Thrombotic Thrombocytopenic Purpura Registry. The annual incidence is estimated at less than 1/1,000,000. Clinical description The majority of patients present during the neonatal period or during childhood but the clinical manifestations are highly variable with mild manifestations of isolated thrombocytopenia throughout childhood in some, and severe neonatal hyperbilirubinemia with episodes of thrombocytopenia and MAHA developing soon after birth in others. In addition, onset may also occur during adulthood, particularly in women when the initial episode of overt TTP is triggered by the first pregnancy.
In these situations, the use of neoadjuvant imatinib can significantly decrease both tumour size and mitotic activity, and permit less radical sphincter-preserving surgery. [25] A substantial proportion of GIST tumors have a high risk of recurrence as estimated by a number of validated risk stratification schemes, and can be considered for adjuvant therapy. [27] [28] The selection criteria underpinning the decision for possible use of imatinib in these settings include a risk assessment based on pathological factors such as tumor size, mitotic rate, and location can be used to predict the risk of recurrence in GIST patients.
Epithelial cell proliferation and fluid secretion that lead to cystogenesis are two hallmark features in ADPKD. [25] During the early stages of cystogenesis, cysts are attached to their parental renal tubules and a derivative of the glomerular filtrate enters the cysts. [19] Once these cysts expand to approximately 2 mm in diameter, the cyst closes off from its parental tubule and after that fluid can only enter the cysts through transepithelial secretion, which in turn is suggested to increase due to secondary effects from an increased intracellular concentrations of cyclic AMP (cAMP). [19] Clinically, the insidious increase in the number and size of renal cysts translates as a progressive increment in kidney volume. [1] [19] Studies led by Mayo Clinic professionals established that the total kidney volume (TKV) in a large cohort of ADPKD patients was 1060 ± 642ml with a mean increase of 204ml over three years, or 5.27% per year in the natural course of the disease, among other important, novel findings that were extensively studied for the first time. [26] Illustration of PKD1 and PKD2 proteins at the cell membrane Diagnosis [ edit ] Usually, the diagnosis of ADPKD is initially performed by renal imaging using ultrasound , CT scan , or MRI . [27] However, molecular diagnostics can be necessary in the following situations: 1- when a definite diagnosis is required in young individuals, such as a potential living related donor in an affected family with equivocal imaging data; [27] 2- in patients with a negative family history of ADPKD, because of potential phenotypic overlap with several other kidney cystic diseases; [27] 3- in families affected by early-onset polycystic kidney disease, since in this cases hypomorphic alleles and/or oligogenic inheritance can be involved; [27] [28] and 4- in patients requesting genetic counseling , especially in couples wishing a pre-implantation genetic diagnosis . [27] [29] The findings of large echogenic kidneys without distinct macroscopic cysts in an infant/child at 50% risk for ADPKD are diagnostic.
Summary Clinical characteristics. Autosomal dominant polycystic kidney disease (ADPKD) is generally a late-onset multisystem disorder characterized by bilateral renal cysts, liver cysts, and an increased risk of intracranial aneurysms. Other manifestations include: cysts in the pancreas, seminal vesicles, and arachnoid membrane; dilatation of the aortic root and dissection of the thoracic aorta; mitral valve prolapse; and abdominal wall hernias. Renal manifestations include hypertension, renal pain, and renal insufficiency. Approximately 50% of individuals with ADPKD have end-stage renal disease (ESRD) by age 60 years. The prevalence of liver cysts increases with age and occasionally results in clinically significant severe polycystic liver disease (PLD).
The fringes of the fimbriae come into contact with the pro-inflammatory follicular fluid. [27] Identical TP53 mutations in STIC and concurrent HGSC were reported, suggesting a clonal relationship. [28] In the triple p53 -PTEN- Dicer knockout mice earlier mentioned, where the Fallopian tubes were intact, HGSC arose from the tubes 100% of the time, eclipsing the tumourgenicity of the ovary. [21] Identical findings were found from PTEN- Dicer knockouts.
These include destabilizing changes in the primary amino acid sequence of the protein, post-translational modifications (such as hyperphosphorylation ), changes in temperature or pH , an increase in production of a protein, or a decrease in its clearance. [1] [5] [15] Advancing age is a strong risk factor, [1] as is traumatic brain injury. [22] [23] In the aging brain, multiple proteopathies can overlap. [24] For example, in addition to tauopathy and Aβ-amyloidosis (which coexist as key pathologic features of Alzheimer's disease), many Alzheimer patients have concomitant synucleinopathy ( Lewy bodies ) in the brain. [25] It is hypothesized that chaperones and co-chaperones (proteins that assist protein folding ) may antagonize proteotoxicity during aging and in protein misfolding-diseases to maintain proteostasis . [26] [27] [28] Seeded induction [ edit ] Some proteins can be induced to form abnormal assemblies by exposure to the same (or similar) protein that has folded into a disease-causing conformation, a process called 'seeding' or 'permissive templating'. [29] [30] In this way, the disease state can be brought about in a susceptible host by the introduction of diseased tissue extract from an afflicted donor.
Prevalence of obesity was more among women (39.5%) than men (14.5%). [23] Japan [ edit ] Using the WHO criteria Japan has the lowest rate of obesity among the OECD member countries at 3.2%. [24] [25] However, as Asian populations are particularly susceptible to the health risks of excess adipose tissue the Japanese have redefined obesity as any BMI greater than 25. [26] Using this cut off value the prevalence of obesity in Japan would be 20%, a threefold increase from 1962 to 2002. [27] A 2008 report stated that 28.6% of men and 20.6% of women in Japan were considered to be obese. [28] Pakistan [ edit ] Main article: Obesity in Pakistan Changing lifestyles, owing to urbanisation, as well as diet issues are the main reasons for obesity in Pakistan.
The curve has flattened in South Africa and Kenya, while Senegal and Equatorial Guinea have seen a steady decline. [27] New strains of the virus were found in December 2020 in South Africa and Nigeria, in addition to the Variant of Concern 202012/01 reported in the United Kingdom in September. [28] The African Union has secured close to 300 million COVID-19 vaccine doses in the largest such agreement yet for Africa, it was announced on January 13, 2021. ... This is 106 deaths per one million population. [58] Niger [ edit ] Main article: COVID-19 pandemic in Niger Niger confirmed its first case on 19 March 2020. [131] Nigeria [ edit ] Main article: COVID-19 pandemic in Nigeria On 27 February, Nigeria confirmed its first case, the first case of coronavirus in sub-Saharan Africa . [132] [133] An Italian citizen who works in Nigeria had returned on 25 February from Milan , Italy through the Murtala Muhammed International Airport , fell ill on 26 February and was transferred to Lagos State Biosecurity Facilities for isolation and testing. [134] [135] [136] The test was confirmed positive by the Virology Laboratory of the Lagos University Teaching Hospital , part of the Nigeria Centre for Disease Control . [137] He was transferred to the Infectious Disease Hospital in Yaba, Lagos . [136] On 28 February, the Lagos State Commissioner for Health announced that the Italian man had travelled on Turkish Airlines with a brief transit at Istanbul . [138] As of 6 March, a total of 219 primary and secondary contacts of the index case had been identified and were being actively monitored. [139] Lock-down measures [ edit ] The Federal government of Nigeria has instructed institutions to shut down for 30 days as a lockdown measure to limit the spread of COVID-19. ... Retrieved 24 March 2020 . ^ "Nigeria confirms first coronavirus case" . BBC News . 28 February 2020. Archived from the original on 2 March 2020 .
HDAg-L, in contrast, is produced during the later stages of an infection, acts as an inhibitor of viral replication, and is required for assembly of viral particles. [28] [29] [30] Thus RNA editing by the cellular enzymes is critical to the virus’ life cycle because it regulates the balance between viral replication and virion assembly. [ citation needed ] Antigenic loop infectivity [ edit ] The HDV envelope protein has three of the HBV surface proteins anchored to it.
Hepatitis delta is a rare hepatic disease characterized by variable degrees of acute hepatitis resulting from infection with the hepatitis delta virus. Occasionally it may present a benign course, but most frequently it manifests with severe liver disease that may include fulminant liver failure, hepatic decompensation and rapid progression to cirrhosis. All patients present concomitant hepatitis B virus infection and an increased risk of developing hepatocellular carcinoma has been reported.
These guidelines stress the use of 28% oxygen masks and caution the dangers of hyperoxia. [ citation needed ] Long-term use of supplemental oxygen improves survival in patients with COPD, but can lead to lung injury. [4] An additional cause of hyperoxia is related to underwater diving with breathing apparatus. ... Barnette Victor Berge Philippe Diolé Gary Gentile Bret Gilliam Bob Halstead Trevor Jackson Steve Lewis John Mattera Rescuers Craig Challen Richard Harris Rick Stanton John Volanthen Frogmen Lionel Crabb Commercial salvors Keith Jessop Science of underwater diving Diving physics Breathing performance of regulators Buoyancy Archimedes' principle Neutral buoyancy Concentration Diffusion Molecular diffusion Force Oxygen fraction Permeation Psychrometric constant Solubility Henry's law Saturation Solution Supersaturation Surface tension Hydrophobe Surfactant Temperature Torricellian chamber Underwater acoustics Modulated ultrasound Underwater vision Snell's law Underwater computer vision Weight Apparent weight Gas laws Amontons's law Boyle's law Charles's law Combined gas law Dalton's law Gay-Lussac's law Ideal gas law Pressure Absolute pressure Ambient pressure Atmospheric pressure Gauge pressure Hydrostatic pressure Metre sea water Partial pressure Diving physiology Artificial gills Cold shock response Diving reflex Equivalent narcotic depth Lipid Maximum operating depth Metabolism Physiological response to water immersion Tissue Underwater vision Circulatory system Blood shift Patent foramen ovale Perfusion Pulmonary circulation Systemic circulation Decompression theory Decompression models: Bühlmann decompression algorithm Haldane's decompression model Reduced gradient bubble model Thalmann algorithm Thermodynamic model of decompression Varying Permeability Model Equivalent air depth Equivalent narcotic depth Oxygen window in diving decompression Physiology of decompression Respiration Blood–air barrier Breathing CO₂ retention Dead space Gas exchange Hypocapnia Respiratory exchange ratio Respiratory quotient Respiratory system Work of breathing Diving environment Classification List of diving environments by type Altitude diving Benign water diving Confined water diving Deep diving Inland diving Inshore diving Muck diving Night diving Open-water diving Black-water diving Blue-water diving Penetration diving Cave diving Ice diving Wreck diving Recreational dive sites Underwater environment Impact Environmental impact of recreational diving Low impact diving Environmental factors Algal bloom Currents: Current Longshore drift Ocean current Rip current Tidal race Undertow Upwelling Ekman transport Halocline Reef Coral reef Stratification Thermocline Tides Turbidity Wind wave Breaking wave Surf Surge Wave shoaling Other Bathysphere Defense against swimmer incursions Diver detection sonar Offshore survey Underwater domain awareness Awards and events Hans Hass Award International Scuba Diving Hall of Fame London Diving Chamber Dive Lectures NOGI Awards Deep-submergence vehicle Aluminaut DSV Alvin American submarine NR-1 Bathyscaphe Archimède FNRS-2 FNRS-3 FNRS-4 Harmony class bathyscaphe Sea Pole -class bathyscaphe Trieste II Deepsea Challenger Ictineu 3 JAGO Jiaolong Konsul -class submersible DSV Limiting Factor Russian submarine Losharik Mir Nautile Pisces -class deep submergence vehicle DSV Sea Cliff DSV Shinkai DSV Shinkai 2000 DSV Shinkai 6500 DSV Turtle DSV-5 Nemo Deep-submergence rescue vehicle LR5 LR7 MSM-1 Mystic -class deep-submergence rescue vehicle DSRV-1 Mystic DSRV-2 Avalon NATO Submarine Rescue System Priz -class deep-submergence rescue vehicle Russian deep submergence rescue vehicle AS-28 Russian submarine AS-34 ASRV Remora SRV-300 Submarine Rescue Diving Recompression System Type 7103 DSRV URF (Swedish Navy) Special interest groups Artificial Reef Society of British Columbia CMAS Europe Coral Reef Alliance Diving Equipment and Marketing Association Divers Alert Network Green Fins Historical Diving Society Karst Underwater Research Nautical Archaeology Program Nautical Archaeology Society Naval Air Command Sub Aqua Club Project AWARE Reef Check Reef Life Survey Rubicon Foundation Save Ontario Shipwrecks SeaKeys Sea Research Society Society for Underwater Historical Research Society for Underwater Technology Underwater Archaeology Branch, Naval History & Heritage Command Submarine escape and rescue Escape trunk International Submarine Escape and Rescue Liaison Office McCann Rescue Chamber Submarine Escape and Rescue system (Royal Swedish Navy) Submarine escape training facility Submarine Escape Training Facility (Australia) Submarine rescue ship Neutral buoyancy facilities for Astronaut training Neutral Buoyancy Laboratory Neutral buoyancy pool Neutral buoyancy simulation as a training aid Neutral Buoyancy Simulator Space Systems Laboratory Yuri Gagarin Cosmonaut Training Center Other Nautilus Productions Category Commons Glossary Indexes: dive sites divers diving Outline Portal
This was discovered using array comparative genomic hybridization , a DNA-based alternative for clinical evaluation of HER2 gene copy number . [28] Trisomy 21 [ edit ] Nuchal edema in Down Syndrome Dr.