Ring chromosome 13 is a chromosomal anomaly of chromosome 13 characterized by a widely variable phenotype (ranging from mild to severe) principally characterized by intrauterine growth retardation, developmental delay, short stature, moderate to severe intellectual deficit, microcephaly, facial dysmorphism (i.e. upslanting palpebral fissures, hypertelorism, abnormal ears, broad nasal bridge, high arched palate, micrognathia, small mouth, and thin lips), hands and feet anomalies, and genital abnormalities. Additional features reported include behavioral problems, hearing and speech disorders, congenital heart defects, cerebral malformations, and anal atresia.
Ring chromosome 13 is a rare chromosome abnormality in which the ends of chromosome 13 join together to form a ring shape. When a ring forms, there may be missing genes where the chromosome's ends fuse together. Therefore, the severity and symptoms associated with ring chromosome 13 vary from person to person, depending on the number of genes, and specific genes, involved. Most people with ring chromosome 13 also have cells with 46 normal chromosomes (this is called mosaicism), which can lessen the effect of the ring chromosome on growth and development. Signs and symptoms that may be present in a person with ring chromosome 13 include developmental delay, slow growth and short stature, feeding difficulties, learning difficulties, a small head size, abnormal formation or positioning of the feet and/or toes, and/or abnormalities of the palate (roof off the mouth).
A developmental anomaly characterized at birth by the presence of right-sided aortic arch, craniofacial dysmorphism (microcephaly, asymmetric, facial bones, broad forehead, borderline hypertelorism, nasal septum deviation, large nasal cavity, large, posteriorly rotated ears, and microstomia with downturned corners), and intellectual disability. These features were observed in 4 members of one family, involving 2 successive generations, suggesting an autosomal dominant mode of transmission. There have been no further descriptions in the literature since 1968.
In a mother and 3 of her children, Strong (1968) found right aortic arch, mental subnormality, and facial peculiarity difficult to describe. Three of the patients had esophageal indentation demonstrated by barium swallow, suggesting left ligamentum arteriosum or anomalous left subclavian artery. Two of the patients had microcephaly. A stillborn child had anencephaly and another died at 10 months with congenital heart disease and microcephaly. Head - Microcephaly Radiology - Esophageal indentation on barium swallow Neuro - Mental retardation Facies - Peculiar facies Inheritance - Autosomal dominant Cardiac - Right aortic arch ▲ Close
A rare genetic neurodegenerative disease characterized by childhood onset of slowly progressive motor and cognitive regression, resulting in intellectual disability and loss of language and ambulation, associated with the appearance of dystonia, parkinsonism, chorea, or rigidity. Ataxia, dysarthria, and seizures have also been reported. Head circumference percentiles may decline over time. Brain imaging shows progressive cerebral and cerebellar atrophy, in some patients also thinning of the corpus callosum.
A number sign (#) is used with this entry because of evidence that childhood-onset neurodegeneration with brain atrophy (CONDBA) is caused by heterozygous mutation in the UBTF gene (600673) on chromosome 17q21. Description CONDBA is a severe progressive neurodegenerative disorder characterized by loss of motor and cognitive skills between ages 2 and 7 years. Affected individuals may have normal development or mild developmental delay, but all eventually lose all motor skills, resulting in inability to walk, absence of language, and profound intellectual disability. Brain imaging shows progressive cerebral and cerebellar atrophy (summary by Edvardson et al., 2017). Clinical Features Edvardson et al. (2017) reported 7 unrelated children, ranging in age from 8 to 23 years, with a childhood-onset neurodegenerative disorder.
Intra-organ variations and other malformations, such as ciliary motricity anomalies (e.g. Kartagener syndrome), biliary atresia and cardiac defects, are frequently associated. Left (polysplenia syndrome) or right (asplenia syndrome) isomerism are usually observed.
See also [ edit ] Situs inversus Situs solitus Chirality (mathematics) Asplenia Polysplenia Ivemark syndrome References [ edit ] ^ "heterotaxy syndrome" . ... PMID 16251896 . ^ a b Kim, Soo-Jin (2011). "Heterotaxy Syndrome" . Korean Circulation Journal . 41 (5): 227–32. doi : 10.4070/kcj.2011.41.5.227 . ... "Splenic Scintigraphy and Radionuclide Venography in the Heterotaxy Syndrome". Radiology . 107 (2): 381–386. doi : 10.1148/107.2.381 . ... "Atrial isomerism in the heterotaxy syndromes with asplenia, or polysplenia, or normally formed spleen: an erroneous concept". ... "Situs Revisited: Imaging of the Heterotaxy Syndrome" . RadioGraphics . 19 (4): 837–852. doi : 10.1148/radiographics.19.4.g99jl31837 .
Heterotaxy is a condition characterized by internal organs that are not arranged as would be expected in the chest and abdomen. Organs are expected to be in a particular orientation inside of the body, known as situs solitus . Heterotaxy occurs when the organs are not in this typical orientation, but are instead in different positions in the body. This most commonly causes complications with the heart, lungs, liver, spleen, and intestines. Specific symptoms include not getting enough oxygen throughout the body, breathing difficulties, increased risk for infection, and problems digesting food.
A rare, genetic, neurological disorder characterized by intrauterine growth retardation, failure to thrive, infantile onset of sensorineural deafness, severe global developmental delay or absent psychomotor development, paraplegia or quadriplegia with dystonia and pyramidal signs, microcephaly, ocular abnormalities (strabismus, optic atrophy), mildly dysmorphic features (deep-set eyes, prominent nasal bridge, micrognathia), seizures and abnormalities of brain morphology (hypomyelinating white matter changes, cerebral atrophy).
Description Deafness, dystonia, and cerebral hypomyelination is an X-linked recessive mental retardation syndrome characterized by almost no psychomotor development, dysmorphic facial features, sensorineural deafness, dystonia, pyramidal signs, and hypomyelination on brain imaging (summary by Cacciagli et al., 2013). Clinical Features Cacciagli et al. (2013) reported 7 males from 3 unrelated families with severe syndromic X-linked mental retardation apparent at birth or in the first years of life and almost no apparent motor or cognitive development. ... Loss of SLC6A8 was consistent with cerebral creatine deficiency syndrome-1 (CCDS1; 300352). Western blot analysis of patient cells showed lack of a normal BCAP31 protein, and Osaka et al. (2012) suggested that the lack of BCAP31 was related to the liver abnormalities and sensorineural deafness observed in their patient. Molecular Genetics In 7 affected males from 3 unrelated families with an X-linked mental retardation syndrome characterized by deafness, dystonia, and central hypomyelination, Cacciagli et al. (2013) identified 3 different hemizygous mutations in the BCAP31 gene (300398.0001-300398.0003).
CADDS is a rare, genetic, neurometabolic disease characterized by severe intrauterine growth retardation, failure to thrive, profound neonatal hypotonia, severe global development delay, elevated very long chain fatty acids in plasma, and neonatal cholestasis leading to hepatic failure and death. Other features include ocular abnormalities (e.g. blindness and cataracts), sensorineural deafness, seizures, and abnormal brain morphology (notably delayed CNS myelination and ventriculomegaly).
A rare genetic skin disease characterized by generalized skin peeling, leukonychia, acral punctate keratoses coalescing into focal keratoderma on the weight-bearing areas, angular cheilitis, and knuckle pads with multiple hyperkeratotic micropapules. The skin appears dry and scaly with superficial exfoliation and underlying erythema. Histopathologic examination of affected skin areas shows hyperkeratosis, acanthosis, and intraepidermal clefting with irregular acantholysis. Additional systemic abnormalities are absent.
Lin et al. (2015) reported 2 unrelated women with peeling skin, leukonychia, acral punctate keratoses, cheilitis, and knuckle pads; the authors proposed the acronym PLACK for the syndrome. The first patient was a 28-year-old Chinese woman who had infantile onset of trauma-induced blistering of the extremities, which worsened in the summer; she also had asymptomatic skin peeling with underlying erythema in the winter.
Cytogenetics Conotruncal heart malformations may be components of certain syndromes, e.g., DiGeorge syndrome (188400), the velocardiofacial syndrome (192430), and genitopalatocardiac syndrome (231060). ... All patients with the deletion showed additional clinical features of the velocardiofacial syndrome. In 7 of the 150 patients (4.6%), the family history was positive for the presence of a conotruncal heart defect. ... His deletion was distal to the usual 3-Mb deletion found in most patients with velocardiofacial syndrome. The deletion did not overlap with any of the previously described 'minimal critical regions' for velocardiofacial syndrome/DiGeorge syndrome. ... The deletion was found to exclude UFD1L (601754), raising questions about the role of this gene in the CATCH22 syndrome. The CDC45L gene (603465) was also excluded from the deletion. ... In a 10-year-old Chinese boy with Langer-Giedion syndrome (150230) and DORV, Tan et al. (2012) identified a de novo balanced chromosomal translocation t(8; 18)(q22;q21) that appeared to disrupt the ZFPM2 gene on chromosome 8q23.
This group of defects is frequently found in patients with 22q11.2 deletion syndrome . A deletion of chromosome 22q11.2 has equally been associated in a subset of patients with various types of isolated non-syndromic conotruncal heart malformations (with the exception of DORV and TGA where this is very uncommon).
Other terms used for Repetitive Strain Injuries are overuse syndrome, musculoskeletal disorders, and cumulative trauma disorders. Some of the more common conditions under these headings include: Cubital Tunnel Syndrome-compression of the ulnar nerve in the cubital tunnel at the elbow.” [6] Pathophysiology [ edit ] In regards to the pathophysiology of ulnar neuropathy:the axon , and myelin can be affected. ... Finally, revisional surgery for cubital tunnel syndrome does not result well for those individuals over 50 years of age. [1] References [ edit ] ^ a b c "Ulnar Nerve Disorders Free Medical Information | Patient" . ... PMID 15965409 . ^ a b "Ulnar Nerve Entrapment at the Elbow (Cubital Tunnel Syndrome)-OrthoInfo - AAOS" . orthoinfo.aaos.org . ... Further reading [ edit ] "NIOSHTIC-2 Publications Search - 20045060 - Do comorbid ulnar symptoms or ulnar neuropathy affect the prognosis of workers with carpal tunnel syndrome?" . www.cdc.gov . Retrieved 2016-07-22 .
You can help by adding to it . ( March 2018 ) [4] [13] [14] See also [ edit ] Vanishing twin Twin-to-twin transfusion syndrome References [ edit ] ^ a b Lopriore, E.; Middeldorp, J.M.; Oepkes, D.; Kanhai, H.H.; Walther, F.J.; Vandenbussche, F.P.H.A. (2007). ... "Prevalence and management of late fetal complications following successful selective laser coagulation of chorionic plate anastomoses in twin-to-twin transfusion syndrome". American Journal of Obstetrics and Gynecology . 194 (3): 796–803. doi : 10.1016/j.ajog.2005.08.069 . ... "Incidence of complications in twin-twin transfusion syndrome after selective fetoscopic laser photocoagulation: a single-center experience". ... "Fetoscopic laser treatment of twin-to-twin transfusion syndrome followed by severe twin anemia-polycythemia sequence with spontaneous resolution". ... "Assessment of Feto-fetal Transfusion Flow Through Placental Arterio-venous Anastomoses in a Unique Case of Twin-to-Twin Transfusion Syndrome". Placenta . 28 (2–3): 209–211. doi : 10.1016/j.placenta.2006.03.006 .
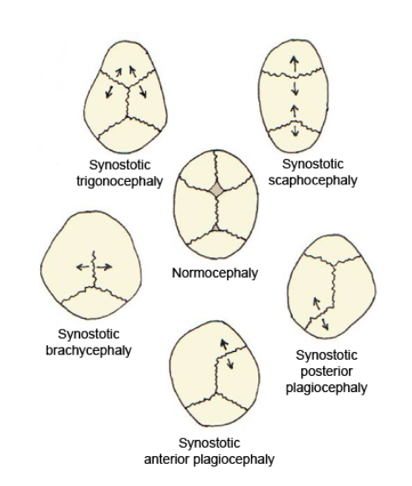
"Flat head syndrome" redirects here. For the condition of an unusually short skull, see Brachycephaly . Plagiocephaly Other names Flat head syndrome Plagiocephaly and other kinds of cranial deformities Specialty Medical genetics Plagiocephaly , also known as flat head syndrome , [1] [2] is a condition characterized by an asymmetrical distortion (flattening of one side) of the skull . ... PMID 20173180 . ^ "Doctor Finds Success In Treating Infants With Flat-Head Syndrome" . CBS Los Angeles. April 30, 2013 . ... "Plagiocephaly and brachycephaly (flat head syndrome) - NHS Choices" . www.nhs.uk . ... External links [ edit ] Classification D ICD - 10 : Q67.3 ICD - 9-CM : 754.0 MeSH : D059041 DiseasesDB : 29858 v t e Congenital malformations and deformations of musculoskeletal system / musculoskeletal abnormality Appendicular limb / dysmelia Arms clavicle / shoulder Cleidocranial dysostosis Sprengel's deformity Wallis–Zieff–Goldblatt syndrome hand deformity Madelung's deformity Clinodactyly Oligodactyly Polydactyly Leg hip Hip dislocation / Hip dysplasia Upington disease Coxa valga Coxa vara knee Genu valgum Genu varum Genu recurvatum Discoid meniscus Congenital patellar dislocation Congenital knee dislocation foot deformity varus Club foot Pigeon toe valgus Flat feet Pes cavus Rocker bottom foot Hammer toe Either / both fingers and toes Polydactyly / Syndactyly Webbed toes Arachnodactyly Cenani–Lenz syndactylism Ectrodactyly Brachydactyly Stub thumb reduction deficits / limb Acheiropodia Ectromelia Phocomelia Amelia Hemimelia multiple joints Arthrogryposis Larsen syndrome RAPADILINO syndrome Axial Skull and face Craniosynostosis Scaphocephaly Oxycephaly Trigonocephaly Craniofacial dysostosis Crouzon syndrome Hypertelorism Hallermann–Streiff syndrome Treacher Collins syndrome other Macrocephaly Platybasia Craniodiaphyseal dysplasia Dolichocephaly Greig cephalopolysyndactyly syndrome Plagiocephaly Saddle nose Vertebral column Spinal curvature Scoliosis Klippel–Feil syndrome Spondylolisthesis Spina bifida occulta Sacralization Thoracic skeleton ribs : Cervical Bifid sternum : Pectus excavatum Pectus carinatum
Dermatofibrosarcoma protuberans (DFSP) is a rare infiltrating soft tissue sarcoma, generally of low grade malignancy, arising from the dermis of the skin and characteristically associated with a specific chromosomal translocation t(17;22). Epidemiology Prevalence is estimated at 1 in 10,000 and annual incidence is estimated at around 1 in 200,000. Clinical description DFSP can present at any age, including infancy and childhood, but usually presents in the 2nd to 5th decade of life. Between 85 and 90% of tumors are low grade lesions, with the remainder classified as the high grade fibrosarcomatous (FS) type. The lesions typically present as an indurated pink or violet-red plaque or nodular mass on the trunk, proximal extremities, or head and neck region.
A number sign (#) is used with this entry because dermatofibrosarcoma protuberans is caused in most cases by a specific fusion of the COL1A1 gene (120150) with the PDGFB gene (190040); see 190040.0002. Description Dermatofibrosarcoma protuberans (DFSP) is an uncommon, locally aggressive, but rarely metastasizing tumor of the deep dermis and subcutaneous tissue. It typically presents during early or middle adult life and is most frequently located on the trunk and proximal extremities (Sandberg et al., 2003). Clinical Features DFSP was first described by Taylor (1890). Sirvent et al. (2003) stated that, because DFSP is relatively rare, grows slowly, and has a low level of aggressiveness, its clinical significance has been underestimated. In particular, they noted that the existence of pediatric cases has been overlooked.
Dermatofibrosarcoma protuberans is an uncommon cancer in which tumors arise in the deeper layers of skin. The tumor usually starts as a small, firm patch of skin; it may be purplish, reddish, or flesh-colored. It is commonly found on the torso, usually in the shoulder and chest area. The tumor typically grows slowly but has a tendency to recur after being removed. It rarely spreads to other parts of the body. The cause of DFSP is unknown, but injury to the affected skin may be a predisposing factor.
ARHGAP31 -, DLL4-, NOTCH1-, and RBPJ- related Adams-Oliver syndrome (AOS) are inherited in an autosomal dominant manner. ... The authors' experience in a tertiary pediatric care center supports a somewhat higher incidence, and further recognition of milder phenotypes within the spectrum of AOS may yet reveal a significantly higher incidence. Differential Diagnosis Syndromic Aplasia Cutis Congenita (ACC) Scalp-ear-nipple (SEN) syndrome (Finlay-Marks syndrome; OMIM 181270). ... DDEB is caused by mutation of COL7A1 . Other causes of syndromic aplasia cutis congenita (ACC) Chromosome disorders Trisomy 13 (Patau syndrome) Wolf-Hirschhorn syndrome (4p- syndrome; OMIM 194190) Setleis syndrome (focal facial dermal dysplasia 3; OMIM 227260), with bitemporal or preauricular skin lesions resembling ACC Johanson-Blizzard syndrome (see Pancreatitis Overview) Oculocerebrocutaneous (Delleman) syndrome (OMIM 164180) Kabuki syndrome Limb body wall complex Knobloch syndrome (OMIM 267750), with high myopia, neuronal elements in scalp defects, occipital encephalocele Various ectodermal dysplasias Isolated aplasia cutis congenita (ACC) Estimated to occur in one in 3,000 live births, most often as an isolated, sporadic malformation [Marneros 2015]. ... The following are examples in which the use of exome sequencing (in clinical practice) facilitated the diagnosis of Adams-Oliver syndrome when other syndromes initially had been suspected and vice versa: One individual with retinopathy of prematurity, small toes, VSD, but no cutis aplasia, was diagnosed initially with Coffin-Siris syndrome. ... This constellation of malformations likely represents a distinct syndrome. Management Evaluations Following Initial Diagnosis To establish the extent of disease and needs in an individual diagnosed with Adams-Oliver syndrome (AOS), the following evaluations are recommended.
Retrieved 10 September 2020 . ^ "F84.5 Asperger syndrome" . World Health Organization . 2015. ... Asperger syndrome or high-functioning autism? . New York: Plenum press. pp. 11–25. ... "The classification of autism, Asperger's syndrome, and pervasive developmental disorder" . ... "Premature popularization of Asperger syndrome" . In Schopler E, Mesibov GB, Kunce LJ (eds.). Asperger syndrome or high-functioning autism? . New York: Plenum press. pp. 388–90.
The disorder should be differentiated from several other conditions, especially centrotemporal spikes without seizures, centrotemporal spikes with local brain pathology, central spikes in Rett syndrome and fragile X syndrome , malignant Rolandic epilepsy , temporal lobe epilepsy and Landau-Kleffner syndrome . [ citation needed ] Treatment [ edit ] Given the benign nature of the condition and the low seizure frequency, treatment is often unnecessary. ... Benign Childhood Partial Seizures and Related Epileptic Syndromes . London: John Libbey Eurotext. pp. 33–100. ... Epileptic Syndromes in Infancy, Childhood, and Adolescence . ... "Panayiotopoulos syndrome: a prospective study of 192 patients" . ... (October 2010). "Panayiotopoulos syndrome: A clinical, EEG, and neuropsychological study of 93 consecutive patients" .
Characteristically, Landau-Kleffner syndrome (LKS) affects previously normal children who undergo a regression of receptive and/or expressive language abilities. ... Huppke et al. (2005) reported an isolated case of Landau-Kleffner syndrome in a boy. Speech development was normal until 2.5 years, when he developed dysarthria. ... Carvill et al. (2013) also reported 2 sisters with Landau-Kleffner syndrome with onset of seizures in early childhood, mild intellectual disability, and speech and language difficulties. ... Lemke et al. (2013) identified heterozygous mutations in the GRIN2A gene (see, e.g., 138253.0005; 138253.0011-138253.0013) in 27 (7.5%) of 359 patients from 2 independent cohorts with idiopathic focal epilepsy syndromes, including Landau-Kleffner syndrome, CSWS, atypical rolandic epilepsy, and benign epilepsy of childhood with centrotemporal spikes (BECTS; 117100). ... By sequence analysis of the GRIN2A gene in 519 probands with a range of epileptic encephalopathies, Carvill et al. (2013) identified heterozygous mutations in 4 probands, all of whom came from the cohort of 44 patients with epilepsy-aphasia syndromes (9% of probands with epilepsy-aphasia syndromes).
Rolandic epilepsy (RE) is a focal childhood epilepsy characterized by seizures consisting of unilateral facial sensory-motor symptoms, with electroencephalogram (EEG) showing sharp biphasic waves over the rolandic region. It is an age-related epilepsy, with excellent outcome. Epidemiology RE is the most common childhood epilepsy and accounts for 8-25% of all childhood epilepsies. Its incidence has been estimated to be approximately 1/5,000 in children within 15 years. Clinical description Onset is between 3 and 12 years, in otherwise normal children (peak of onset is 5-8 years). Seizures typically occur during sleep or drowsy states; they are brief with unilateral sensorimotor (such as numbness, tingling, drooling) that involves pharynx, tongue, face, lips and sometimes hand.
Description Benign epilepsy of childhood with centrotemporal spikes (BECTS) or sharp waves, also known as rolandic epilepsy, is the most common idiopathic childhood epilepsy syndrome (Neubauer et al., 1998). It is termed 'rolandic' epilepsy because of the characteristic features of partial seizures involving the region around the lower portion of the central gyrus of Rolando.
Camurati-Engelmann disease is a skeletal condition that is characterized by abnormally thick bones (hyperostosis) in the arms, legs, and skull. The thick limb bones can lead to bone pain and muscle weakness in the arms and legs and cause individuals with Camurati-Engelmann disease to tire quickly. Bone pain ranges from mild to severe and can increase with stress, activity, or cold weather. Leg weakness can make it difficult to stand up from a seated position and some affected individuals develop a waddling or unsteady walk. Additional limb abnormalities include joint deformities (contractures), knock knees, and flat feet (pes planus ).
Camurati-Englemann disease (CED) is a rare, clinically variable bone dysplasia syndrome characterized by hyperostosis of the long bones, skull, spine and pelvis, associated with severe pain in the extremities, a wide-based waddling gait, joint contractures, muscle weakness and easy fatigability. Camurati-Englemann disease (CED) is a rare, clinically variable bone dysplasia syndrome characterized by hyperostosis of the long bones, skull, spine and pelvis, associated with severe pain in the extremities, a wide-based waddling gait, joint contractures, muscle weakness and easy fatigability. ... Disorders to consider include craniodiaphyseal dysplasia, autosomal dominant Kenny-Caffey syndrome, juvenile Paget disease, Ghosal hematodiaphyseal dysplasia, Worth type autosomal dominant osteosclerosis, sclerosteosis and hyperostosis corticalis generalisata (see these terms).
The sclerosis of the long bones in CDD is restricted to the diaphyses; in CED metaphyses can also be affected. Kenny-Caffey syndrome type 2 (OMIM 127000) FAM111A AD Sclerosis of long bones, cortical thickening, medullary stenosis Hypocalcemia, hypoparathyroidism, delayed fontanelle closure Juvenile Paget disease (OMIM 239000) TNFRSF11B AR Cranial hyperostosis, sensorineural hearing loss, sclerosis of long bones Predisposition to fractures, bowing of the long bones Ghosal hematodiaphyseal dysplasia (OMIM 231095) TBXAS1 AR Diaphyseal sclerosis Severe anemia; leukopenia & thrombocytopenia Endosteal hyperostosis, AD (OMIM 144750) LRP5 AD Diaphyseal sclerosis (endosteal), cranial nerve involvement in some Wide deep mandible w/↑ gonial angle (distinct from the enlarged mandible found only occasionally in CED) SOST -related sclerosing bone dysplasias incl sclerosteosis & van Buchem disease SOST AR Cranial hyperostosis, cranial nerve involvement, diaphyseal sclerosis Syndactyly, dysplastic or absent nails AD = autosomal dominant; AR = autosomal recessive; MOI = mode of inheritance Management Evaluations Following Initial Diagnosis To establish the extent of disease and needs in an individual diagnosed with Camurati-Engelmann disease (CED), the initial evaluation should include the following if they have not already been completed: Complete skeletal survey Assessment for cranial nerve deficits, including neurologic examination, audiology evaluation, and ophthalmologic evaluation Baseline blood pressure if considering treatment with losartan CBC to evaluate for anemia in individuals with significant endosteal involvement If acute bone pain is present, consideration of serum ESR and bone scan examination as baseline measures of disease activity In individuals with radiographic evidence of skull base sclerosis and neurologic symptoms, consideration of baseline CT examination of the head and neck to determine the extent of disease and allow consideration of surgical treatment options Consultation with a clinical geneticist and/or genetic counselor Treatment of Manifestations No consensus management guidelines have been developed to date. ... Losartan has an anti-TGFβ effect and is being tested in individuals with Marfan syndrome. Treatment with losartan has not improved bone pain in some individuals [Yuldashev et al 2017] Other analgesics and non-pharmacologic methods are frequently used for alleviation of pain. ... Because the mechanism of CED involves increased TGFB1 signaling, also found in Marfan syndrome and Loeys-Dietz syndrome, this death is of some concern.
Camurati-Engelmann disease is a genetic condition that mainly affects the bones. People with this disease have increased bone density, particularly affecting the long bones of the arms and legs. In some cases, the skull and hip bones are also affected. The thickened bones can lead to pain in the arms and legs, a waddling walk, muscle weakness, and extreme tiredness. The age that symptoms begin varies greatly, but most people with this condition develop pain or weakness by adolescence. Camurati-Engelmann disease is caused by a mutation in the TGFB1 gene and inheritance is autosomal dominant.
A number sign (#) is used with this entry because of evidence that Camurati-Engelmann disease results from domain-specific heterozygous mutations in the transforming growth factor-beta-1 gene (TGFB1; 190180) on chromosome 19q13. Also see Camurati-Engelmann disease type 2 (606631) in which no mutation in the TGFB1 gene has been found. Description Camurati-Engelmann disease is a rare autosomal dominant type of bone bone dysplasia. The hallmark of the disorder is the cortical thickening of the diaphyses of the long bones. Hyperostosis is bilateral and symmetrical and usually starts at the diaphyses of the femora and tibiae, expanding to the fibulae, humeri, ulnae, and radii.