-
Familial Thoracic Aortic Aneurysm
Wikipedia
There is an association between familial thoracic aortic aneurysm and Marfan syndrome as well as other hereditary connective tissue disorders. ... External links [ edit ] Classification D ICD - 9-CM : 441.00 OMIM : 607086 DiseasesDB : 30073 External resources eMedicine : emerg/28 GeneReview/NCBI/NIH/UW entry on Thoracic Aortic Aneurysms and Aortic Dissections v t e Cardiovascular disease (vessels) Arteries , arterioles and capillaries Inflammation Arteritis Aortitis Buerger's disease Peripheral artery disease Arteriosclerosis Atherosclerosis Foam cell Fatty streak Atheroma Intermittent claudication Critical limb ischemia Monckeberg's arteriosclerosis Arteriolosclerosis Hyaline Hyperplastic Cholesterol LDL Oxycholesterol Trans fat Stenosis Carotid artery stenosis Renal artery stenosis Other Aortoiliac occlusive disease Degos disease Erythromelalgia Fibromuscular dysplasia Raynaud's phenomenon Aneurysm / dissection / pseudoaneurysm torso : Aortic aneurysm Abdominal aortic aneurysm Thoracic aortic aneurysm Aneurysm of sinus of Valsalva Aortic dissection Aortic rupture Coronary artery aneurysm head / neck Intracranial aneurysm Intracranial berry aneurysm Carotid artery dissection Vertebral artery dissection Familial aortic dissection Vascular malformation Arteriovenous fistula Arteriovenous malformation Telangiectasia Hereditary hemorrhagic telangiectasia Vascular nevus Cherry hemangioma Halo nevus Spider angioma Veins Inflammation Phlebitis Venous thrombosis / Thrombophlebitis primarily lower limb Deep vein thrombosis abdomen Hepatic veno-occlusive disease Budd–Chiari syndrome May–Thurner syndrome Portal vein thrombosis Renal vein thrombosis upper limb / torso Mondor's disease Paget–Schroetter disease head Cerebral venous sinus thrombosis Post-thrombotic syndrome Varicose veins Gastric varices Portacaval anastomosis Caput medusae Esophageal varices Hemorrhoid Varicocele Other Chronic venous insufficiency Chronic cerebrospinal venous insufficiency Superior vena cava syndrome Inferior vena cava syndrome Venous ulcer Arteries or veins Angiopathy Macroangiopathy Microangiopathy Embolism Pulmonary embolism Cholesterol embolism Paradoxical embolism Thrombosis Vasculitis Blood pressure Hypertension Hypertensive heart disease Hypertensive emergency Hypertensive nephropathy Essential hypertension Secondary hypertension Renovascular hypertension Benign hypertension Pulmonary hypertension Systolic hypertension White coat hypertension Hypotension Orthostatic hypotension This cardiovascular system article is a stub .MYLK, NOTCH1, FBN1, SMAD3, COL3A1, FOXE3, LOX, SRFBP1, ACTA2, MYH11, TGFBR2, AAT1, ACTB, MMP3, PPARG, EFEMP2

-
Fanconi Anemia
Orphanet
Congenital malformations may involve other organ systems and vary within families. Short stature is syndromic and/or associated to endocrinopathies. ... Differential diagnosis FA clinical manifestations overlap with many malformation syndromes (Dubowitz, Seckel, Holt-Oram, Baller-Gerold, thrombocytopenia-absent radius, Nijmegen breakage syndromes, VACTERL association, dyskeratosis congenital, Blackfan-Diamond anemia) and diagnosis of FA is often delayed until a patient develops BMF or malignancies. FA should be considered in the differential diagnosis of all young patients with BMF of unknown etiology, and in other cancer predisposition syndromes (Bloom, Rothmund-Thomson or Werner syndromes) or syndromes with either constitutional or acquired BMF.FANCC, FANCG, BRIP1, FANCM, FANCA, FANCD2, SLX4, FANCL, RAD51C, RAD51, BRCA1, ERCC4, UBE2T, XRCC2, MAD2L2, BRCA2, FANCI, FANCF, FANCB, FANCE, PALB2, TNF, ERCC1, RFWD3, MX1, USP1, ZNF276, FAH, MUL1, POLG, CHEK1, GAS2, FAN1, PRKN, CBLL2, ATM, TP53, BLM, IFNG, FUT1, CD34, ATR, PARP1, NBN, RAD18, RUNX1, COL11A2, FAAP20, BACH1, ALDH2, SNU13, RPS19, PCNA, DLK1, H2AX, FAAP24, DCLRE1A, MRE11, NLRP2, MLH1, HPRT1, SPTAN1, RAD50, MSH2, WDR48, UCN, HES1, IL6, E2F1, PTEN, GYPA, HNF4A, HSP90AA1, COX4I1, IFNA1, IFNA13, CSF3, GH1, DCLRE1B, OCRL, NPM1, IL3, IGF1, IL1B, IL2, TP53BP1, HPGDS, TXN, SYF2, ME1, RAD51D, KAT5, TGFB1, ABL1, RB1, PRTN3, MAPK1, PPARG, SCT, XRCC3, POLD1, PMS2, RNF8, MSC, PAK1, OPRM1, PRDX6, SMARCA4, SOD2, STAT1, TERC, KITLG, MMP9, ERVW-4, UHRF1, GATA3, CASP3, OPN4, CDK1, CDC42, FAAP100, ATAD5, CTBP1, CTSB, DDX11, TIMM8A, NLN, ERCC2, MECOM, TTK, MKS1, REV1, MSH6, GGH, LOH19CR1, XRCC5, ZNF236, LEPQTL1, CPNE3, CXCR4, AIMP2, USP11, MTDH, BRAP, BMF, FAM120B, ARHGAP24, TRAF2, CUL4B, DONSON, PPM1D, USP48, CDK10, RUVBL1, FSD1L, TNFSF10, BCL2L12, ZBTB32, XRCC4, RBM45, EZR, DEL11P13, FECD3, XRCC6P5, MIR34A, TYRO3, MIR181C, UBE2B, MIR146A, GSTK1, RRDX, VCAM1, WEE1, SLCO6A1, WNT5A, CENPX, WRN, RNF168, WT1, BABAM1, XPA, ROMO1, XPC, XPO1, NDUFAF6, TLR7, NEIL1, CT55, USP16, EDIL3, SLC12A9, UIMC1, ABCC4, IKZF1, RETN, RACK1, PAG1, ATF7IP, IL17RB, AHSA1, PLK2, GINS2, RUVBL2, PPARGC1A, FSTL1, CHEK2, MCM10, DKK1, BRMS1, RNF19A, NEIL3, PARPBP, SETBP1, PTPRU, DNM1L, PHGDH, EFCAB6, MBD2, FSD1, EXO1, XPR1, ELOVL6, MAPKAPK2, GPR132, TLR8, GRAP2, WNK1, MRPL36, POLDIP2, PCBP4, ZBTB7A, TBPL1, GORASP1, PKNOX2, VPS9D1, UBL5, HDAC9, TELO2, SALL4, SH2B3, TIGAR, RECQL4, AAVS1, TPO, FXN, FOXF1, FHIT, FEN1, EZH2, ERCC5, ERBB2, EPO, EPHB2, ENO1, EIF4E, EGR1, DNA2, NQO1, DHFR, DAXX, FN1, FRA16D, CYP2B6, MTOR, GYPE, GYPB, GSTT1, GSTP1, GSTM1, GSK3B, GPX4, GOLGA4, GLRX, GFAP, GABRG1, GABRB1, XRCC6, FUS, FRZB, CYP11A1, CYP2A6, HIC1, CCNB1, RUNX3, CAV1, CAT, CASP8, BIK, BCS1L, BCR, BCL6, BAX, ATRX, AR, ALDH1A1, AFP, ADH5, ACP1, CCK, CCNE1, CTNNB1, CD33, CTNS, CSF2, MAPK14, CRK, CREBBP, ABCC2, CCR7, CLCN5, CFL1, CDKN1A, CDH1, CD68, CD48, CD44, CD38, HBG2, HIF1A, TP73, RNF4, RELA, REG1A, OPN1LW, RBBP8, RAD52, RAD51B, RAC1, PTCH1, PSMA7, PSEN1, EIF2AK2, MAPK8, PRKDC, PRKAA2, PMS1, REV3L, ATXN1, PHF1, CCL3, TOP1, TH, TERF2, ZEB1, TBX1, TAP2, SYT1, STAT5B, STAT5A, SPTBN1, SPP1, SOAT1, SLC1A3, SFRP1, SELP, PIN1, SLC25A3, HLA-DRB1, MEF2C, MCM6, MCM4, SMAD4, LRP2, LPL, STMN1, KIT, JAK2, ITGB1, INSR, INHBA, IL10, IL3RA, ICAM1, HSPA4, MECP2, MEN1, PFKFB3, MGMT, PDGFRB, CHMP1A, PAX6, NOTCH1, NME1, NCAM1, MYH4, MYH2, MTHFR, MSH3, MRC1, MMP7, MMP2, KMT2A, CIITA, PSPH

-
Mandibulofacial Dysostosis With Ptosis, Autosomal Dominant
Omim
Genetic analysis excluded linkage to the TCOF1 gene (606847) on chromosome 5q, mutation in which causes Treacher Collins syndrome (154500). The authors distinguished the syndrome in this family from other disorders with similar phenotypes, including Goldenhar syndrome (164210) and the Bauru type of mandibulofacial dysostosis (604830).
-
Santos Syndrome
Omim
Clinical Features Santos et al. (2008) reported 6 members of a Brazilian family with short stature due to fibular agenesis or hypoplasia, clubfeet with oligodactyly, acromial dimples, motion limitations of the forearms and/or hands, and severe nail hypoplasia or anonychia that was sometimes associated with brachydactyly and occasionally with preaxial polydactyly. Inheritance Santos et al. (2008) suggested autosomal dominant inheritance with incomplete penetrance for this disorder based on the structure of the pedigree, but noted that autosomal recessive inheritance could not be ruled out because of the high rate of consanguinity (about 20-30%) in the region to which the family belongs. INHERITANCE - Autosomal dominant GROWTH Height - Short stature SKELETAL Limbs - Acromial dimples - Limited extension, flexion, pronation and/or supination of the forearm and/or hand - Fibular agenesis/hypoplasia - Hypoplastic femora - Asymmetric lower limbs - Genua valga Hands - Brachydactyly - Preaxial polydactyly - Postaxial polydactyly - Syndactyly - Hypoplastic distal phalanx - Swan neck defect of the finger Feet - Oligodactyly - Clubfeet - Talipes equinovarus - Pes metatarsus varus SKIN, NAILS, & HAIR Nails - Anonychia - Ungual hypoplasia ▲ Close
-
Granddad Syndrome
Omim
Clinical Features Marion et al. (1989) observed 7 patients who presented a prematurely aged facial appearance and the following features: intrauterine growth retardation with postnatal growth delay, normal mental development, and decreased subcutaneous fat. The facial appearance included triangular facies, prominent forehead, thin or absent scalp hair, deep-set eyes, midfacial hypoplasia, prominent nasal septum with hypoplasia of the alae nasi, prominent ears, and thin lips. Inheritance Marion et al. (1989) suggested autosomal dominant inheritance of this disorder because 3 of the patients were from the same family--2 daughters and their father. Neuro - Normal mental development Facies - Prematurely aged facial appearance - Triangular facies - Prominent forehead - Deep-set eyes - Midfacial hypoplasia - Prominent nasal septum - Hypoplastic alar nasae - Prominent ears - Thin lips Inheritance - Autosomal dominant Hair - Thin or absent scalp hair Growth - Intrauterine growth retardation - Postnatal growth delay Skin - Decreased subcutaneous fat ▲ Close
-
Congenital Disorder Of Glycosylation
Wikipedia
POMT1/POMT2-CDG ( Walker-Warburg syndrome and Muscle-Eye-Brain syndrome )) with deficiencies in O -mannosylation of proteins; O -xylosylglycan synthesis defects (EXT1/EXT2-CDG ( hereditary multiple exostoses ) and B4GALT7-CDG ( Ehlers-Danlos syndrome , progeroid variant)); O -fucosylglycan synthesis (B3GALTL-CDG (Peter's plus syndrome) and LFNG-CDG ( spondylocostal dysostosis III)). ... "Carbohydrate-deficient glycoprotein syndrome type V: deficiency of dolichyl-P-Glc:Man9GlcNAc2-PP-dolichyl glucosyltransferase" . ... "Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy" . ... "Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome)". Nature Genetics . 16 (1): 88–92. doi : 10.1038/ng0597-88 . ... "Carbohydrate deficient glycoprotein syndrome type II: a deficiency in Golgi localised N-acetyl-glucosaminyltransferase II" .PMM2, SRD5A3, MPI, SLC35A2, MOGS, DPM1, DOLK, MPDU1, SLC35A1, FCSK, ALG1, MAGT1, SRD5A3-AS1, TMEM165, PGM1, COG8, ALG6, COG6, ALG3, ALG9, PGM3, ALG8, DPAGT1, MAN1B1, COG7, COG5, APOC3, MGAT2, ALG12, DPM3, SLC35C1, SERPINA1, LEMD3, ALPI, ALPP, ATP6V0A2, ALG11, SLC39A8, STT3B, MAN1A1, GOLPH3, CD47, STT3A, IAPP, DHDDS, RFT1, ALG2, PGAP3, NUS1P3, COG2, GFM1, SLC35A3, POFUT2, CCDC115, NUS1, PGAP1, ALG13, CSGALNACT1, TRAPPC11, GRIN3A, AGA, RXYLT1, IGF1, GPI, B4GALT1, FUT8, FXN, F11, SLC26A3, DLD, DDOST, DCN, DAG1, SERPINA6, BGN, BCHE, ATP6AP1, SERPINC1, APRT, APOB, HP, IGFBP5, MGAM, ITGB2, ST3GAL5, DPM2, TUSC3, AKR1B1, TF, SSR4, SSR3, ST3GAL3, SI, RNASE4, RAB1A, PYCR1, ATXN3, MFAP1, LAMP2, LAMP1, LAD1, PGR-AS1
-
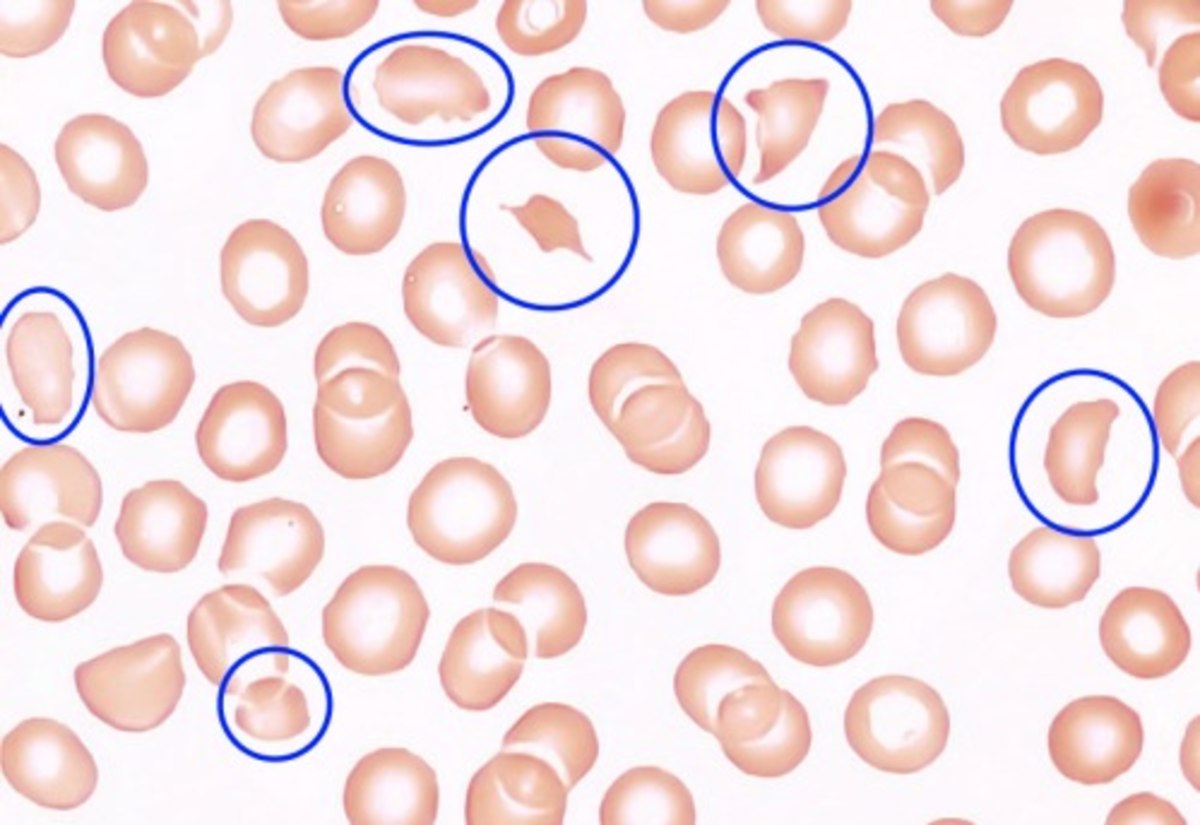
Upshaw–schulman Syndrome
Wikipedia
Upshaw–Schulman syndrome Blood smear under the microscope with typical schistocytes in TTP marked in blue – H&E stain Specialty Rheumatology Upshaw–Schulman syndrome ( USS ) is the recessively inherited form of thrombotic thrombocytopenic purpura (TTP), a rare and complex blood coagulation disease. ... A deficiency of ADAMTS13 activity in first-degree relatives is also a very strong indicator for an Upshaw-Schulman syndrome. [1] [2] [9] Treatment [ edit ] The therapy of an acute TTP episode has to be started as early as possible. [1] [2] The standard treatment is the daily replacement of the missing ADAMTS13 protease in form of plasma infusions or in more severe episodes by plasma exchange . ... "Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes" . Hematology . 2012 : 604–609. doi : 10.1182/asheducation.V2012.1.604.3798564 . ... "The incidence of thrombotic thrombocytopenic purpura — hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency" . ... "Decreased cold-insoluble globulin in congenital thrombocytopenia (Upshaw-Schulman syndrome)". New England Journal of Medicine . 300 (7): 368. doi : 10.1056/nejm197902153000717 .ADAMTS13, THBD, F3, TFPI, VWF, CFH, HLA-DRB1, THBS1, ZFP36, TNFSF10, MAPKAPK2, ABCA1, PTPN22, TNS3, IL33, RBM45, KCNH8, RN7SL263P, SH3BP4, CCL2, STAT3, APOA1, S100B, PTGS2, PHEX, COX2, JAK2, HP, BRF1, FLT4, EPHB1, ELAVL1, SLC25A10, CASP1, CACNA1S, MTCO2P12
-
X-Linked Otopalatodigital Spectrum Disorders
Gene_reviews
The X-linked otopalatodigital (X-OPD) spectrum disorders, characterized primarily by skeletal dysplasia, include the following: Otopalatodigital syndrome type 1 (OPD1) Otopalatodigital syndrome type 2 (OPD2) Frontometaphyseal dysplasia type 1 (FMD1) Melnick-Needles syndrome (MNS) Terminal osseous dysplasia with pigmentary skin defects (TODPD) In OPD1, most manifestations are present at birth; females can present with severity similar to affected males, although some have only mild manifestations. ... DD = developmental delay; FMD1 = frontometaphyseal dysplasia type 1; GU = genitourinary; MNS = Melnick-Needles syndrome; OPD1 = otopalatodigital syndrome type 1; OPD2 = otopalatodigital syndrome type 2; SNHL = sensorineural hearing loss; TODPD = terminal osseous dysplasia with pigmentary skin defects 1. ... In males, the spectrum of severity ranges from mild manifestations in otopalatodigital syndrome type 1 (OPD1) to a more severe presentation in frontometaphyseal dysplasia type 1 (FMD1) and otopalatodigital syndrome type 2 (OPD2). Prenatal lethality is the only clinical phenotype described in males with Melnick-Needles syndrome (MNS) [Spencer et al 2018]. Females exhibit variable expressivity. ... OPD1 was also called Taybi syndrome after its first description in 1963.
-
Benzodiazepine Withdrawal Syndrome
Wikipedia
Many doctors do not adjust benzodiazepine dosage according to age in elderly patients. [158] See also [ edit ] Psychiatry portal Alcohol withdrawal syndrome Benzodiazepine dependence Benzodiazepine equivalence Opioid withdrawal syndrome Physical dependence Post-acute-withdrawal syndrome Rebound effect SSRI discontinuation syndrome Neuroleptic discontinuation syndrome References [ edit ] ^ a b c d e f Soyka M (2017). ... "Benzodiazepine dependence: Focus on withdrawal syndrome". Annales Pharmaceutiques Françaises . 67 (6): 408–13. doi : 10.1016/j.pharma.2009.07.001 . ... "Sevrage en benzodiazépines révélé par un syndrome douloureux abdominal pseudochirurgical" [Benzodiazepine withdrawal masquerading as surgical abdominal syndrome]. ... PMID 2070121 . ^ Mellor, CS; Jain, VK (1982). "Diazepam withdrawal syndrome: Its prolonged and changing nature" . ... PMID 2300914 . ^ De Bard, ML (1979). "Diazepam withdrawal syndrome: A case with psychosis, seizure, and coma".

-
Post-Concussion Syndrome
Wikipedia
S2CID 38741663 . ^ a b King NS. 2003. Post-concussion syndrome: clarity amid the controversy? ... PMID 10798893 . ^ a b c Evans RW (1992). "The postconcussion syndrome and the sequelae of mild head injury". ... "Traumatic brain injury and the post-concussion syndrome: A diffusion tensor tractography study" . ... "Diagnostic criteria for postconcussional syndrome after mild to moderate traumatic brain injury" . ... "Predicting postconcussion syndrome after minor traumatic brain injury".
-
Obstructive Sleep Apnea
Wikipedia
Some of these craniofacial syndromes are genetic, others are from unknown causes. In many craniofacial syndromes, the features that are unusual involve the nose, mouth, and jaw, or resting muscle tone, and put the individual at risk for OSA syndrome. Down syndrome is one such syndrome. In this chromosomal abnormality, several features combine to make the presence of obstructive sleep apnea more likely. ... Obstructive sleep apnea does occur even more frequently in people with Down syndrome than in the general population. ... Cleft palate syndromes are such an example. During the newborn period, all humans are obligate nasal breathers .EDNRA, EDN1, ACE, IL1B, IL10, ADORA1, GHSR, GHRL, ABCB1, MTOR, SLC18A3, SNAP25, BMP2, CCDC162P, SRCAP, MECP2, CHAT, EP300, UNC80, MAP6, SYT2, CCDC47, SELENON, CDH4, TRPV4, HRAS, NOTCH2, ADIPOQ, SLC5A7, NT5E, COL2A1, PLCB4, COL6A1, COL13A1, TSPAN18, CREBBP, CRP, KMT2A, NONO, AUTS2, DNA2, TRIM8, FGFR3, CENPJ, PCGF2, REM1, REN, GNAI3, SCN8A, IL6, H3-3A, SH3BP2, AHDC1, AGTPBP1, SLC25A1, IGF1, VAMP1, LINC02770, IDUA, IDS, TNF, APOE, LEP, PLCB1, SMCO2, CACNA1A, MYO9A, NGLY1, AGRN, ASXL2, QRICH1, MAGEL2, ARRB1, PSG5, ARCN1, POGZ, SLC35A2, NFIX, LEPR, HTR2A, COPD, VEGFA, SLC6A4, MMP9, HIF1A, NOS3, CD274, ASAP2, PDAP1, TUSC2, PAPOLA, ASAP1, REG3A, MRPS30, MTPAP, CLOCK, CXCL8, HCRT, ACP3, CCL2, TLR4, TXN, DECR1, CYBA, CHI3L1, GPT, SETD2, TLR2, ARSA, AGT, LAMC2, LBP, CST3, BGLAP, EPO, CYP2D6, ATP6AP2, CSF2, SERPINE1, BDNF, CAD, PER1, NOS1, VDR, SOD1, IFNG, PPARG, PLA2G15, NGF, TAC1, IL4, NR3C2, SHBG, TGFB1, ADAMTS2, VCAM1, GRAP2, TNFRSF11B, NFE2L2, TSPAN31, LGALS3, PDCD1, ESM1, IL18, TPO, IRS1, PTGS2, SELENBP1, MIF, STIN2-VNTR, STAT3, MIR145, GABPA, GABBR1, ACSS2, FABP4, AMY1C, CD40LG, MIR34A, NGB, MIR107, NANS, DBP, ARNTL, SESN2, ACCS, CRY1, IL33, CHIT1, CHGA, CCHCR1, RETN, HMOX1, FOXP3, HP, ALB, GSN, NRG1, AGER, AMY1B, HMGB1, AMY1A, VIP, VWF, ENHO, MCIDAS, NANOS1, IL1R2, SYT1, ZFP36, TEK, PLA2G7, MIRLET7D, AIMP2, C1QTNF9, ARID1A, TNFSF11, RNMT, IL18R1, FFAR4, ABCC8, SERPINA12, APLN, SULT1A3, PHOX2B, LOC107987479, SUMO1, MTCO2P12, MIR130A, TERC, TFF3, TFRC, PRDX2, TG, TACR1, MIR664A, TGM2, MIR596, MIR574, TH, ECSCR, TIMP1, FIQTL1, NPS, TM7SF2, PLF, MICOS10-NBL1, TNNI3, MIR26B, MIR200B, TTN, PGR-AS1, PPARGC1B, POLDIP2, RBM45, ADAM22, FASTK, SDS, APOM, ADAM29, CD160, CAPN10, SOX21, DCTN3, ANGPTL8, CHP1, SP140, PHB2, DYM, AHSP, SLC33A1, ANGPTL4, SIRT1, SENP1, CPS1-IT1, MMD, FAM155B, B3GAT1, SLC17A5, FLRT2, STAR, PANX1, PART1, RNF19A, MAN1C1, ARID1B, CFAP97, HPSE, DCD, XPR1, KL, APOA5, NLRP3, UCN3, FGF21, ADAMTS4, OPN4, ADAMTS3, ABCC11, COL18A1, DNAJC5, MAP9, HERPUD1, CAMKMT, PCLAF, ABI1, EBI3, CEBPZ, FTO, PROCR, AHSA1, RAI1, WNK1, GORASP1, HAMP, ROCK2, ABCA1, SREBF1, FPR1, FLNC, FOXO3, F2R, ETV3, ESRRA, ESR1, EREG, EPHB1, ELN, ELK3, ELAVL2, EGFR, EGF, EDNRB, LPAR1, DYNC1H1, DMP1, CTNNB1, CSPG4, FN1, FPR2, CSH1, FPR3, HOXD13, HLA-DRB1, HLA-DQB1, HLA-B, HAL, GZMB, NR3C1, GPD1, GJA5, GJA1, GHRH, GHR, GDNF, GCH1, GCG, GC, LRRC32, GAP43, GAD1, CSH2, CSF3, HSPA1A, VPS51, ATM, AR, FAS, KLK3, APOH, APOB, BIRC5, XIAP, BIRC3, APCS, ANXA5, ANGPT2, ANGPT1, ALOX5, ALDOA, AGTR1, JAG1, ADRB3, ADCYAP1, C3, CA2, MAPK14, SLC25A20, CRY2, CRK, CPB2, COMT, COL3A1, CCR5, CLU, ERCC8, CEACAM5, CDKN2B, CDKN2A, CD36, CD34, CD19, KRIT1, CAV1, CALM3, CALM2, CALM1, HES1, HSPA1B, SRI, PTGS1, PTGER3, PTGDS, PSPH, MAPK1, PRKAB1, PRKAA2, PRKAA1, PRB1, POU2AF1, PON1, PNMT, PLEK, PITX3, PIK3CG, PIK3CD, ACP5, PIK3CA, PGF, PF4, PTH, PTX3, PAPPA, RARRES2, SRC, SNCG, SNCB, SLC6A2, SLC5A2, SLC2A4, SGCD, SFTPC, SFRP5, SFRP1, SELE, CX3CL1, CXCL5, SCD, S100A8, ROCK1, RNASE2, RHAG, RELA, SLC26A4, OSM, HSPA4, SMAD7, SH2D1A, LTBP3, LTB, LPL, LPA, LCN2, KCNMB1, KCNK3, KCNJ2, KCNJ1, ITGA2B, ISG20, IRF1, INSR, IL2RA, IGFALS, ICAM1, HSPG2, HSPA9, SMAD3, MC4R, OPRM1, MCL1, NUCB2, NTRK1, NPR3, NPR2, NPY, NOS2, NHS, NBL1, MYH7, MTHFR, COX2, COX1, MPZ, MMP7, MMP2, MMP1, MGP, MET, MEOX2, PIK3CB

-
Campylobacteriosis
Wikipedia
Campylobacteriosis Other names Campylobacter food poisoning – campylobacter enteritis [1] Specialty Infectious disease Symptoms Diarrhea, abdominal pain, fever Complications toxic megacolon , dehydration , sepsis , Guillain–Barré syndrome Causes Campylobacter jejuni Treatment Supportive care , Antibiotics (select cases) Prognosis Usually self-limited Deaths Infrequent Campylobacteriosis is an infection by the Campylobacter bacterium , [2] most commonly C. jejuni . ... It produces an inflammatory, sometimes bloody, diarrhea or dysentery syndrome, mostly including cramps, fever and pain. ... The effect is known as an acute idiopathic demyelinating polyneuropathy (AIDP), i.e. Guillain–Barré syndrome, in which one sees symptoms of ascending paralysis, dysaesthesias usually below the waist, and, in the later stages, respiratory failure. [ citation needed ] Some strains of C jejuni produce a cholera -like enterotoxin, which is important in the watery diarrhea observed in infections. ... In a small number of cases, the infection may be associated with hemolytic uremic syndrome and thrombotic thrombocytopenic purpura through a poorly understood mechanism. [ citation needed ] Transmission [ edit ] The common routes of transmission for the disease-causing bacteria are fecal-oral, person-to-person sexual contact, ingestion of contaminated food (generally unpasteurized (raw) milk and undercooked or poorly handled poultry ), and waterborne (i.e., through contaminated drinking water ). ... External links [ edit ] Campylobacter jejuni genomes and related information at PATRIC , a Bioinformatics Resource Center funded by NIAID Classification D ICD - 10 : A04.5 ICD - 9-CM : 008.43 MeSH : D002169 DiseasesDB : 1914 External resources MedlinePlus : 000224 eMedicine : ped/2697 med/263 v t e Proteobacteria -associated Gram-negative bacterial infections α Rickettsiales Rickettsiaceae / ( Rickettsioses ) Typhus Rickettsia typhi Murine typhus Rickettsia prowazekii Epidemic typhus , Brill–Zinsser disease , Flying squirrel typhus Spotted fever Tick-borne Rickettsia rickettsii Rocky Mountain spotted fever Rickettsia conorii Boutonneuse fever Rickettsia japonica Japanese spotted fever Rickettsia sibirica North Asian tick typhus Rickettsia australis Queensland tick typhus Rickettsia honei Flinders Island spotted fever Rickettsia africae African tick bite fever Rickettsia parkeri American tick bite fever Rickettsia aeschlimannii Rickettsia aeschlimannii infection Mite-borne Rickettsia akari Rickettsialpox Orientia tsutsugamushi Scrub typhus Flea-borne Rickettsia felis Flea-borne spotted fever Anaplasmataceae Ehrlichiosis : Anaplasma phagocytophilum Human granulocytic anaplasmosis , Anaplasmosis Ehrlichia chaffeensis Human monocytotropic ehrlichiosis Ehrlichia ewingii Ehrlichiosis ewingii infection Rhizobiales Brucellaceae Brucella abortus Brucellosis Bartonellaceae Bartonellosis : Bartonella henselae Cat-scratch disease Bartonella quintana Trench fever Either B. henselae or B. quintana Bacillary angiomatosis Bartonella bacilliformis Carrion's disease , Verruga peruana β Neisseriales M+ Neisseria meningitidis/meningococcus Meningococcal disease , Waterhouse–Friderichsen syndrome , Meningococcal septicaemia M− Neisseria gonorrhoeae/gonococcus Gonorrhea ungrouped: Eikenella corrodens / Kingella kingae HACEK Chromobacterium violaceum Chromobacteriosis infection Burkholderiales Burkholderia pseudomallei Melioidosis Burkholderia mallei Glanders Burkholderia cepacia complex Bordetella pertussis / Bordetella parapertussis Pertussis γ Enterobacteriales ( OX− ) Lac+ Klebsiella pneumoniae Rhinoscleroma , Pneumonia Klebsiella granulomatis Granuloma inguinale Klebsiella oxytoca Escherichia coli : Enterotoxigenic Enteroinvasive Enterohemorrhagic O157:H7 O104:H4 Hemolytic-uremic syndrome Enterobacter aerogenes / Enterobacter cloacae Slow/weak Serratia marcescens Serratia infection Citrobacter koseri / Citrobacter freundii Lac− H2S+ Salmonella enterica Typhoid fever , Paratyphoid fever , Salmonellosis H2S− Shigella dysenteriae / sonnei / flexneri / boydii Shigellosis , Bacillary dysentery Proteus mirabilis / Proteus vulgaris Yersinia pestis Plague / Bubonic plague Yersinia enterocolitica Yersiniosis Yersinia pseudotuberculosis Far East scarlet-like fever Pasteurellales Haemophilus : H. influenzae Haemophilus meningitis Brazilian purpuric fever H. ducreyi Chancroid H. parainfluenzae HACEK Pasteurella multocida Pasteurellosis Actinobacillus Actinobacillosis Aggregatibacter actinomycetemcomitans HACEK Legionellales Legionella pneumophila / Legionella longbeachae Legionnaires' disease Coxiella burnetii Q fever Thiotrichales Francisella tularensis Tularemia Vibrionaceae Vibrio cholerae Cholera Vibrio vulnificus Vibrio parahaemolyticus Vibrio alginolyticus Plesiomonas shigelloides Pseudomonadales Pseudomonas aeruginosa Pseudomonas infection Moraxella catarrhalis Acinetobacter baumannii Xanthomonadaceae Stenotrophomonas maltophilia Cardiobacteriaceae Cardiobacterium hominis HACEK Aeromonadales Aeromonas hydrophila / Aeromonas veronii Aeromonas infection ε Campylobacterales Campylobacter jejuni Campylobacteriosis , Guillain–Barré syndrome Helicobacter pylori Peptic ulcer , MALT lymphoma , Gastric cancer Helicobacter cinaedi Helicobacter cellulitis v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum v t e Chicken Rooster Hen Chick As poultry Chicken as food List of chicken dishes Breeds Capon Poularde Poussin Pullet Broiler Husbandry Battery cage Free range Furnished cages Yarding Chicken tractor Poultry farming Broiler industry Beak trimming Hatchery Chick sexing Chick culling Candling Abnormal behaviour of birds in captivity European Union Council Directive 1999/74/EC Culture Chickens as pets Cockatrice Cockfighting Kapparos Sarimanok Rooster Flag Diseases Aspergillosis Avian infectious laryngotracheitis Avian influenza Avian sarcoma leukosis virus Histomoniasis (blackhead disease) Botulism Campylobacteriosis Candidiasis Coccidiosis Colds Dermanyssus gallinae Egg binding Erysipelas Fatty liver hemorrhagic syndrome Fowlpox Gallid alphaherpesvirus 3 Gapeworm Infectious bursal disease Infectious coryza in chickens Marek's disease Mycoplasmas Newcastle disease Omphalitis Psittacosis Pullorum Scaly leg Squamous cell carcinoma Tibial dyschondroplasia Toxoplasmosis

-
Hereditary Gingival Fibromatosis
Wikipedia
It can cover teeth in various degrees, and can lead to aesthetic disfigurement. [2] Fibrous enlargement is most common in areas of maxillary and mandibular tissues of both arches in the mouth . [1] Phenotype and genotype frequency of HGF is 1:175,000 where males and females are equally affected but the cause is not entirely known. [2] [3] It mainly exists as an isolated abnormality but can also be associated with a multi-system syndrome. [1] Contents 1 Signs and symptoms 1.1 Obvious signs 2 Cause 2.1 Genetic 2.2 Non genetic 3 Mechanism 4 Diagnosis 5 Prevention 6 Treatment 6.1 If left untreated 6.2 Treatment 7 Recent research 8 See also 9 References 10 External links Signs and symptoms [ edit ] There may or may not be any evidence of history of HGF in the family nor any usage of taking long-term medicines for any particular disease when it comes to diagnosing HGF. ... Inflammation [3] [7] Hormonal Imbalance [3] [7] Neoplasia [3] [7] More commonly associated with an autosomal dominant gene inheritance [2] Multi-system syndromes: Zimmerman-Laband syndrome , Jones syndrome , Ramon syndrome , Rutherford syndrome , juvenile hyaline fibromatosis , systemic infantile hyalinosis , and mannosidosis [8] Some unknown causes [3] [7] Mechanism [ edit ] Genetic linkage studies are among the most popular methods of study to look at the mechanism of this HGF. ... However, in some cases where it can develop as a result of rare multi-system syndromes, such as: Zimmerman-Laband, Jones, Ramon Syndrome, Rutherford Syndrome, Juvenile Hyaline Fibromatosis, Systemic Infantile Hyalinosis, and Mannosidosis , it is best for one to simply monitors the possible progression for HGF with regular dental check-ups. [8] If the patient's disease is treated by means of surgery, it is recommended that the patient undergoes post-surgical therapies for maintenance and periodic monitoring of gums for the sake of the possibility of re-occurrence of HGF. [8] Treatment [ edit ] This disease has not been shown to be life-threatening or the cause of death in patients. ... This case study also acknowledged how HGF can be part of a multi-system syndrome associated with disorders such as Zimmermann Laband syndrome (ear, nose, bone, and nail defects with hepatosplenomegaly), Rutherford syndrome (microphthalmia, mental retardation, athetosis, and hypopigmentation), Murray-Puretic Drescher syndrome and Ramon syndrome . [1] See also [ edit ] Medicine portal Chronic periodontitis Epidemiology of periodontal diseases Gingivitis Gum graft Periodontist Tooth loss Gingival recession References [ edit ] ^ a b c d e f g h i j k l m n o p q r Poulami Majumder, Vineet Nair, Malancha Mukherjee, Sujoy Ghosh, and Subrata Kumar Dey, “The Autosomal Recessive Inheritance of Hereditary Gingival Fibromatosis,” Case Reports in Dentistry, vol. 2013, Article ID 432864, 4 pages, 2013. doi:10.1155/2013/432864 ^ a b c d e f g h i j Thomas C. ... Sarigelou, “Current concepts on gingival fibromatosis-related syndromes,” Journal of Investigative and Clinical Dentistry, vol. 2, no. 3, pp. 156–161, 2011.SOS1, GINGF2, REST, HGF, TGFB1, MET, IL6, TIMP1, SERPINH1, FN1, VEGFA, IL1B, HPSE, MMP1, EGFR, MMP2, CCN2, CAV1, TGFBR2, TGFB3, THAS, TGFB2, TIMP2, SMN2, SMN1, SHH, TNF, TP53, ACTB, SOCS1, SLC52A2, LINC02605, COMETT, GINGF3, MIR335, MIR93, NRK, PWAR1, ASH1L, NR1I2, SLC25A37, UNC50, PAK4, ENAM, SEMA3E, CD163, MAPK8, MSC, PIK3CA, MAPK1, HBG1, GPT, FGF1, F2R, EZH2, EPHB2, MARK2, EGF, CTNNB1, CDKN2A, CDK9, CDK2, BMP7, BMP2, ASCL1, XIAP, HAS3, HBG2, PIK3R1, HPN, PIK3CG, PIK3CD, PIK3CB, ALB, PCNA, NGF, MMP9, KISS1, IL13, CXCL8, IL7, IGF1, IFNG, ICAM1, HES1, H3P10
-
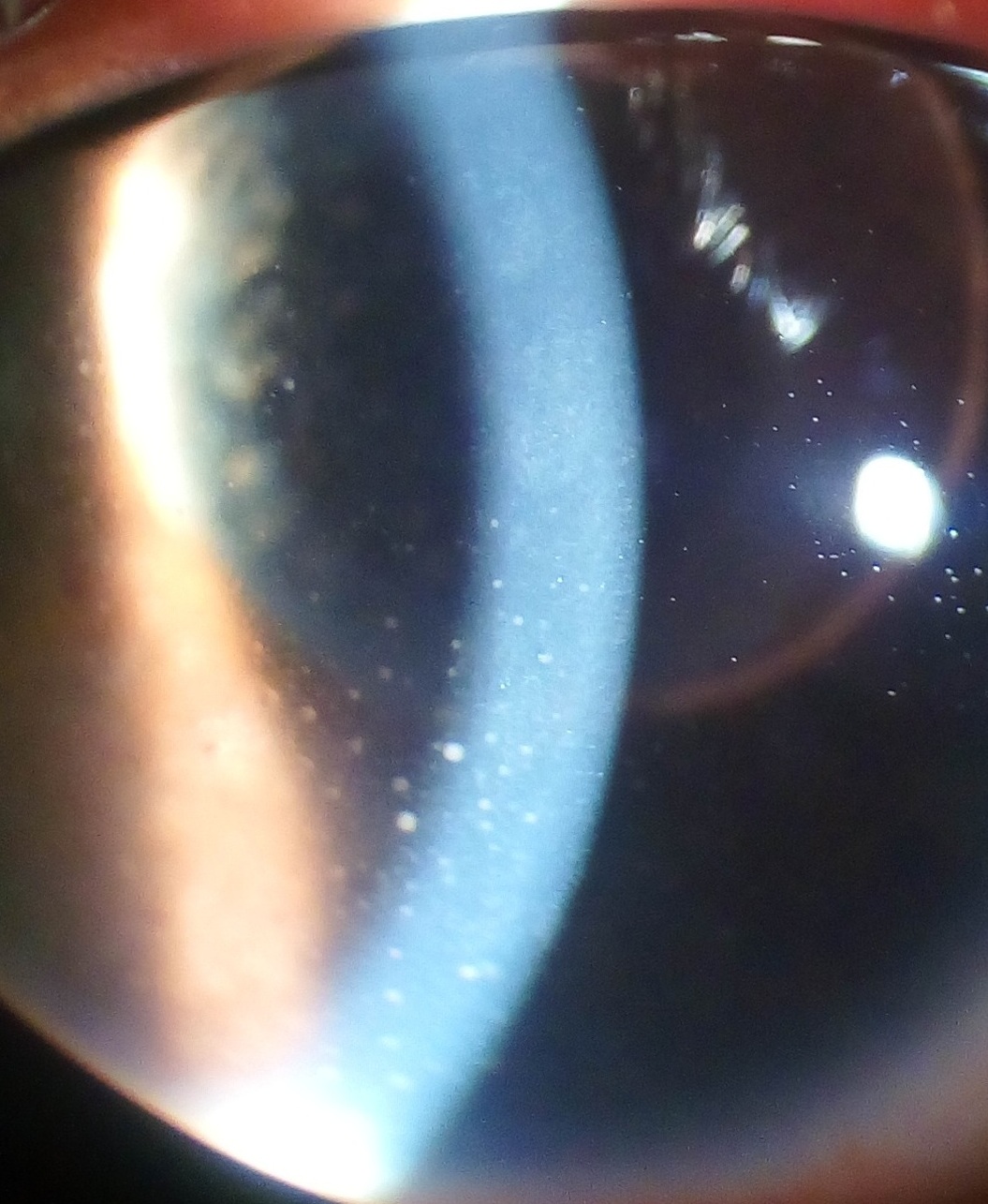
Uveitis
Wikipedia
Please consider expanding the lead to provide an accessible overview of all important aspects of the article. ( January 2014 ) Uveitis Inflammation of the eye and keratic precipitates due to uveitis Pronunciation [ˌju.vi.ˈai.tɪs] Specialty Ophthalmology Symptoms Headaches, red eyes, blurred vision, photophobia, burning and redness of the eye Causes Behçet disease , Crohn's disease , Fuchs heterochromic iridocyclitis , Granulomatosis with polyangiitis , HLA-B27 related uveitis, Juvenile idiopathic arthritis , Sarcoidosis , Spondyloarthritis , Sympathetic ophthalmia , Tubulointerstitial nephritis and uveitis syndrome , brucellosis , herpesviruses , leptospirosis , Lyme disease , presumed ocular histoplasmosis syndrome , syphilis , toxocariasis , toxoplasmic chorioretinitis , tuberculosis , Zika fever Uveitis is the inflammation of the uvea , the pigmented layer that lies between the inner retina and the outer fibrous layer composed of the sclera and cornea . ... Contents 1 Signs and symptoms 1.1 Anterior uveitis (iritis) 1.2 Intermediate uveitis 1.3 Posterior uveitis 2 Causes 2.1 Noninfectious or autoimmune causes 2.2 Infectious causes 2.3 Associated with systemic diseases 2.4 Drug related side effects 2.5 White Dot syndromes 2.6 Masquerade syndromes 3 Pathophysiology 3.1 Immunologic factors 3.2 Genetic factors 3.3 Infectious agents 4 Diagnosis 4.1 Classification 5 Treatment 6 Prognosis 7 Epidemiology 8 See also 9 References 10 External links Signs and symptoms [ edit ] Ciliary flush Hypopyon in anterior uveitis, seen as yellowish exudate in lower part of anterior chamber of eye Anterior uveitis (iritis) [ edit ] Burning of the eye Redness of the eye Blurred vision Photophobia Irregular pupil Signs of anterior uveitis include dilated ciliary vessels , presence of cells and flare in the anterior chamber, and keratic precipitates ("KP") on the posterior surface of the cornea . ... In anterior uveitis, no associated condition or syndrome is found in approximately one-half of cases. However, anterior uveitis is often one of the syndromes associated with HLA-B27 . Presence of this type of HLA allele has a relative risk of evolving this disease by approximately 15%. [3] The most common form of uveitis is acute anterior uveitis (AAU). ... In some of these cases, the presentation in the eye is characteristic of a described syndrome, which are called white dot syndromes , and include the following diagnoses: acute posterior multifocal placoid pigment epitheliopathy birdshot chorioretinopathy multifocal choroiditis and panuveitis multiple evanescent white dot syndrome punctate inner choroiditis serpiginous choroiditis acute zonal occult outer retinopathy Masquerade syndromes [ edit ] Masquerade syndromes are those conditions that include the presence of intraocular cells but are not due to immune-mediated uveitis entities.RBP3, SERPINF1, HSPA9, CALB2, ALB, TNFRSF1A, CRYBB2, TNF, SOD2, CRYAA, SAG, IL6, IL17A, IL1B, IL1A, CCL2, TLR4, ICAM1, HLA-DQB1, HLA-DQA1, IL18, CX3CL1, CXCL10, ARG1, CCL5, ANXA1, NR3C1, CX3CR1, NOD2, RELA, VIP, TJP1, CCR5, KCNJ10, ASS1, ICOS, AQP4, AQP1, SNCB, AOC3, HLA-DRB1, CAPN5, NLRP3, FASLG, FAS, NLRP1, RASGRP1, TCF4, MST1, GPR35, PRKCD, MBTPS2, NLRC4, IKBKG, BTNL2, DNASE1L3, CASP10, IL10, HLA-B, IFNG, HLA-A, IL27, IL23R, CRP, RBM45, ACE, CFH, MYD88, MIR155, IL2, IL17D, MIR146A, IL22, KIR3DL1, AIRE, SERPINA1, CD274, MIR223, MC5R, TLR2, TGFB2, SIRT1, MICA, AIF1, PDCD1, AHR, MIR21, FOXP3, IFNA1, S100A12, IGAN1, S100A8, CDR3, IFNA2, IFNA13, CFI, MYDGF, THBD, IL1RN, CYP27B1, CXCR4, VEGFA, SOCS5, SLC9A3R2, KLF4, GRAP2, TM7SF2, AIMP1, SCAF11, SOCS3, SOCS1, TRAF5, AIMP2, TRAF1, GEMIN2, BAS, KNTC1, ADM, ZEB2, TRIM69, ERAP2, RTN4R, VTCN1, TLR10, KIAA1109, ADO, DNER, HT, DPYSL5, TICAM1, IFNL2, SUMO4, C1orf141, MIR30B, MIR326, PSS, CXCL16, MRAP, PTPRU, POLDIP2, CXCL13, AHSA1, BTG3, SMUG1, PLA2G15, ARIH1, RNF19A, ACAD8, ATF7IP, TRIB2, IL19, TBX21, MZB1, TRPV2, ERAP1, EIM, IL23A, POMC, TGFBR3, KIR2DS1, EGF, EGR2, EPRS1, ESD, ETS1, EXTL3, GABPA, GC, GEM, HLA-C, HMGB1, HMOX1, HSPA1L, IFNB1, IGH, IL1RAP, IL6ST, CXCL8, IL15, ATN1, DECR1, DBP, BRAF, AKR1B1, AMD1, AMD1P2, ANGPT1, ANXA11, APCS, XIAP, OPN1SW, C3AR1, CYP24A1, CD14, CD40, CKB, CISH, CRK, MAPK14, VCAN, CTLA4, IRF5, KIR3DL2, TRBV20OR9-2, LCN2, ACAN, PPIA, MAPK1, MAPK3, MAP2K7, PSMB9, PTEN, PTPN2, PTPRF, REG1A, RLBP1, RPE, S100A1, S100B, CCL8, CCL13, CCL20, STAT1, STAT4, PLXNA2, PIK3CG, PIK3CD, MS, SH2D1A, EPCAM, MAP6, MBL2, CD46, NR3C2, MOG, MPO, ND4, PIK3CB, MTX1, NFE2L2, NOS2, OVGP1, P2RX7, PFDN5, SLC25A3, PIK3CA, LINC-ROR
-
Pendred Syndrome/nonsyndromic Enlarged Vestibular Aqueduct
Gene_reviews
The clinical diagnosis of Pendred syndrome is established in a proband with SNHL, characteristic temporal bone abnormalities identified on thin-cut CT, and euthyroid goiter. ... Diagnosis Suggestive Findings The diagnosis of Pendred syndrome/nonsyndromic enlarged vestibular aqueduct (PDS/NSEVA) spectrum is suggested by the following clinical, temporal bone imaging, and endocrine findings. ... Clinical Characteristics Clinical Description Pendred syndrome/nonsyndromic enlarged vestibular aqueduct (PDS/NSEVA) comprises a phenotypic spectrum of sensorineural hearing loss (SNHL), vestibular dysfunction, and temporal bone abnormalities. ... Although enlarged vestibular aqueduct (EVA) with or without cochlear hypoplasia are seen in virtually all individuals with Pendred syndrome (PDS), neither EVA nor cochlear hypoplasia is specific for PDS. Other causes of these types of temporal bone malformations include congenital cytomegalovirus and branchiootorenal syndrome, in which there is no associated thyroid abnormality.
-
22q11.2 Deletion Syndrome
Gene_reviews
Individuals with 22q11.2 deletion syndrome (22q11.2DS) can present with a wide range of features that are highly variable, even within families. ... Diagnosis Suggestive Findings 22q11.2 deletion syndrome (22q11.2DS) should be suspected in individuals with the following clinical findings. ... Genes of Interest in the Differential Diagnosis of 22q11.2 Deletion Syndrome View in own window Gene(s) Disorder MOI Key Clinical Features Overlapping w/22q11.2 Deletion Syndrome CHD7 CHARGE syndrome AD CHD, palatal anomalies, coloboma, choanal atresia, growth deficiency, ear anomalies / hearing loss, DDs, facial palsy, genitourinary anomalies, & immunodeficiency DHCR7 Smith-Lemli-Opitz syndrome AR Polydactyly & cleft palate JAG1 NOTCH2 Alagille syndrome AD Butterfly vertebrae, CHD, & posterior embryotoxon TBX1 1 Tetralogy of Fallot (OMIM 187500) AD CHD, preauricular pits AD = autosomal dominant; AR = autosomal recessive; CHARGE = c oloboma, h eart defects, choanal a tresia, r estricted growth and development, g enital abnormalities, and e ar anomalies; CHD = congenital heart disease; DDs = developmental delays; MOI = mode of inheritance 1. ... Oculoauriculovertebral (Goldenhar) syndrome (OAVS) (when ear anomalies, vertebral defects, heart disease, renal anomalies are present) (OMIM 141400) Teratogenic exposures. ... Management Evaluations Following Initial Diagnosis Clinical practice guidelines for the evaluation and treatment of individuals with 22q11.2 deletion syndrome (22q11.2DS) have been published.TBX1, COMT, CRKL, DGCR, HIRA, UFD1, DGCR2, DGCR8, DGCR6, FGF8, GP1BB, RREB1, ESS2, JMJD1C, ARVCF, SEC24C, CHRD, TRAPPC10, MAPK1, PRICKLE1, EDNRA, PRODH, GSC2, MED15, TBX5, MRPL40, PRDM9, MBD5, DGCR6L, GCOM1, SCZD12, POLR2M, ARSA, SNAP29, APOL1, CLDN5, TBX3, SLC25A1, SLC7A4, RANBP1, SEPTIN5, HTC2, EDN1, CLTC, CHRNA7, MYZAP
-
Periventricular Nodular Heterotopia 1
Omim
The disorder in 3 boys with PVNH, cerebellar hypoplasia, severe mental retardation, epilepsy, and syndactyly was designated the BPNH/MR syndrome (Fink et al., 1997). Guerrini and Dobyns (1998) described a 'new' syndrome of BPNH with frontonasal malformation and mild mental retardation in 2 unrelated boys, aged 8 and 5.5 years. ... The 5.5-year-old patient had several additional features of Aarskog syndrome (100050), including shawl scrotum and cryptorchidism. The combination of widow's peak and shawl scrotum has also been reported in autosomal dominant Teebi hypertelorism (145420), autosomal recessive Aarskog-like faciodigitogenital syndrome (227330), and X-linked Aarskog-Scott syndrome (305400). ... A gene-dosage model for BPHN/MR is supported by observations in boys with the X-Y(Xq) syndrome (Lahn et al., 1994). The X-Y(Xq) syndrome results from aberrant meiotic exchange between Xq and Yq in the fathers, which produces translocation of a portion of distal Xq28 to the Y chromosome inherited by each boy. ... Hehr et al. (2006) noted that the initial working diagnosis made in this patient was cerebrofrontofacial syndrome (see 243310). Unger et al. (2007) suggested that the patient reported by Hehr et al. (2006) may have had FG syndrome-2 (FGS2; 300321), given his constipation and dysmorphic facial features.ERMARD, FLNA, ARFGEF2, NEDD4L, MAP1B, ARF1, TMTC3, DCHS1, FMR1, FAT4, ZSWIM6, MAN1B1, RTTN, C2CD3, KAT6B, ANKRD11, SLC25A24, DHCR7, CSF1R, PAFAH1B1, DCX, SYBU, PHF10, FAM20C, INTS8, RMDN3, KIAA0319L, MOB2, RMDN2, PLEKHG6, AKT1, DCDC2, RMDN1, DLL1, SMUG1, NES, HESX1, YWHAE, RELN, MEN1, MAP3K4, LLGL1, GNAI2, ERN1, ARX
-
Bile Acid Malabsorption
Wikipedia
Most people with previous ileal resection and chronic diarrhea will have abnormal SeHCAT tests and can benefit from bile acid sequestrants . [ citation needed ] People with primary bile acid diarrhea are frequently misdiagnosed as having irritable bowel syndrome . [13] When SeHCAT testing is performed, the diagnosis of primary bile acid diarrhea is commonly made. In a review of 18 studies of the use of SeHCAT testing in diarrhea-predominant irritable bowel syndrome patients, 32% of 1223 people had a SeHCAT 7-day retention of less than 10%, and 80% of these reported a response to cholestyramine, a bile acid sequestrant . [12] Estimates of the population prevalence suggest that 1% of the adult population could have primary bile acid diarrhea (Type 2 bile acid malabsorption). [12] References [ edit ] ^ a b c Bannaga, A; Kelman, L; O'Connor, M; Pitchford, C; Walters, JR; Arasaradnam, RP (2017). ... "Systematic review: the prevalence of idiopathic bile acid malabsorption as diagnosed by SeHCAT scanning in patients with diarrhoea-predominant irritable bowel syndrome" . Aliment. Pharmacol. Ther . 30 (7): 707–17. doi : 10.1111/j.1365-2036.2009.04081.x . ... S2CID 3454888 . ^ Hofmann, AF (1967). "The syndrome of ileal disease and the broken enterohepatic circulation: cholerheic enteropathy" . ... External links [ edit ] Classification D ICD - 10 : K90.8 ICD - 9-CM : 579.8 OMIM : 613291 MeSH : C567652 DiseasesDB : 6650 v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum
-
Excited Delirium
Wikipedia
Syndrome that presents with multiple, etiologies including psychomotor agitation and delirium Excited delirium Other names Excited delirium syndrome, agitated delirium, Sudden death in restraint syndrome An example of physical restraints which may be used until chemical sedation takes effect. ... (November 2012). "Excited Delirium Syndrome (ExDS): defining based on a review of the literature" . ... "Review, clinical update, and practice guidelines for excited delirium syndrome". Journal of Special Operations Medicine . 15 (1): 62–9. ... "Excited Delirium and Sudden Death: A Syndromal Disorder at the Extreme End of the Neuropsychiatric Continuum" . ... "ACEP Recognizes Excited Delirium as Unique Syndrome". Emergency Medicine News . 31 (11): 4. doi : 10.1097/01.EEM.0000340950.69012.8d .

-
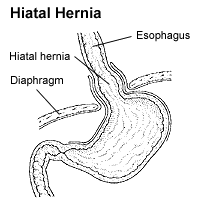
Hiatal Hernia
Wikipedia
However surgery has its own risks including death and disability, so that even for large or paraesophageal hernias, watchful waiting may on balance be safer and cause fewer problems than surgery. [12] Complications from surgical procedures to correct a hiatal hernia may include gas bloat syndrome , dysphagia (trouble swallowing), dumping syndrome , excessive scarring, and rarely, achalasia . [12] [13] Surgical procedures sometimes fail over time, requiring a second surgery to make repairs. ... External links [ edit ] Classification D ICD - 10 : K44 , Q40.1 ICD - 9-CM : 553.3 , 750.6 OMIM : 142400 MeSH : D006551 DiseasesDB : 29116 External resources MedlinePlus : 001137 eMedicine : med/1012 radio/337 Wikimedia Commons has media related to Hiatal hernia . v t e Diseases of the digestive system Upper GI tract Esophagus Esophagitis Candidal Eosinophilic Herpetiform Rupture Boerhaave syndrome Mallory–Weiss syndrome UES Zenker's diverticulum LES Barrett's esophagus Esophageal motility disorder Nutcracker esophagus Achalasia Diffuse esophageal spasm Gastroesophageal reflux disease (GERD) Laryngopharyngeal reflux (LPR) Esophageal stricture Megaesophagus Esophageal intramural pseudodiverticulosis Stomach Gastritis Atrophic Ménétrier's disease Gastroenteritis Peptic (gastric) ulcer Cushing ulcer Dieulafoy's lesion Dyspepsia Pyloric stenosis Achlorhydria Gastroparesis Gastroptosis Portal hypertensive gastropathy Gastric antral vascular ectasia Gastric dumping syndrome Gastric volvulus Buried bumper syndrome Gastrinoma Zollinger–Ellison syndrome Lower GI tract Enteropathy Small intestine ( Duodenum / Jejunum / Ileum ) Enteritis Duodenitis Jejunitis Ileitis Peptic (duodenal) ulcer Curling's ulcer Malabsorption : Coeliac Tropical sprue Blind loop syndrome Small bowel bacterial overgrowth syndrome Whipple's Short bowel syndrome Steatorrhea Milroy disease Bile acid malabsorption Large intestine ( Appendix / Colon ) Appendicitis Colitis Pseudomembranous Ulcerative Ischemic Microscopic Collagenous Lymphocytic Functional colonic disease IBS Intestinal pseudoobstruction / Ogilvie syndrome Megacolon / Toxic megacolon Diverticulitis / Diverticulosis / SCAD Large and/or small Enterocolitis Necrotizing Gastroenterocolitis IBD Crohn's disease Vascular : Abdominal angina Mesenteric ischemia Angiodysplasia Bowel obstruction : Ileus Intussusception Volvulus Fecal impaction Constipation Diarrhea Infectious Intestinal adhesions Rectum Proctitis Radiation proctitis Proctalgia fugax Rectal prolapse Anismus Anal canal Anal fissure / Anal fistula Anal abscess Hemorrhoid Anal dysplasia Pruritus ani GI bleeding Blood in stool Upper Hematemesis Melena Lower Hematochezia Accessory Liver Hepatitis Viral hepatitis Autoimmune hepatitis Alcoholic hepatitis Cirrhosis PBC Fatty liver NASH Vascular Budd–Chiari syndrome Hepatic veno-occlusive disease Portal hypertension Nutmeg liver Alcoholic liver disease Liver failure Hepatic encephalopathy Acute liver failure Liver abscess Pyogenic Amoebic Hepatorenal syndrome Peliosis hepatis Metabolic disorders Wilson's disease Hemochromatosis Gallbladder Cholecystitis Gallstone / Cholelithiasis Cholesterolosis Adenomyomatosis Postcholecystectomy syndrome Porcelain gallbladder Bile duct / Other biliary tree Cholangitis Primary sclerosing cholangitis Secondary sclerosing cholangitis Ascending Cholestasis / Mirizzi's syndrome Biliary fistula Haemobilia Common bile duct Choledocholithiasis Biliary dyskinesia Sphincter of Oddi dysfunction Pancreatic Pancreatitis Acute Chronic Hereditary Pancreatic abscess Pancreatic pseudocyst Exocrine pancreatic insufficiency Pancreatic fistula Other Hernia Diaphragmatic Congenital Hiatus Inguinal Indirect Direct Umbilical Femoral Obturator Spigelian Lumbar Petit's Grynfeltt-Lesshaft Undefined location Incisional Internal hernia Richter's Peritoneal Peritonitis Spontaneous bacterial peritonitis Hemoperitoneum Pneumoperitoneum v t e Congenital malformations and deformations of digestive system Upper GI tract Tongue , mouth and pharynx Cleft lip and palate Van der Woude syndrome tongue Ankyloglossia Macroglossia Hypoglossia Esophagus EA/TEF Esophageal atresia: types A, B, C, and D Tracheoesophageal fistula: types B, C, D and E esophageal rings Esophageal web (upper) Schatzki ring (lower) Stomach Pyloric stenosis Hiatus hernia Lower GI tract Intestines Intestinal atresia Duodenal atresia Meckel's diverticulum Hirschsprung's disease Intestinal malrotation Dolichocolon Enteric duplication cyst Rectum / anal canal Imperforate anus Rectovestibular fistula Persistent cloaca Rectal atresia Accessory Pancreas Annular pancreas Accessory pancreas Johanson–Blizzard syndrome Pancreas divisum Bile duct Choledochal cysts Caroli disease Biliary atresia Liver Alagille syndrome Polycystic liver diseaseATP7A, COL1A1, GPHN, WDR4, AFG3L2, SPECC1L, NIPBL, TPRKB, OSGEP, NUP133, NUP107, PORCN, SLC2A10, TUBB6, ATAD1, WDR73, TP53RK, ADAMTS2, SLC6A5, LAGE3, PTCH1, COL5A1, COL5A2, TNXB, GLRB, GLRA1, ALDH18A1, SMARCB1, TCF4, TGFB3, CHST14, IL12B, HLA-DMA, GER, FUT3, ECI1, ADGRV1, CYP2C19, COL3A1