Etiology This embryopathy appears between the 5th and 12th week of pregnancy due to a failure in the fusion of the frontal processes (fronto-nasal process, medial and lateral nasal processes, maxillary process). Cleft lip is an isolated, non-syndromic anomaly in 70% of cases. The remaining 30% of cases are seen in at least 300 syndromes where cleft lip is just one of the featured anomalies. Non-syndromic clefts are possibly caused by a combination of genetic and environmental factors. ... Differential diagnosis The presence of associated malformations allows for differentiation between isolated and syndromic forms. Antenatal diagnosis Antenatal diagnosis is often possible with a prenatal ultrasound.
Low blood sugar , low blood pressure , hyponatremia , hypercapnia , hypoxia, slowed heart rate , and hypoventilation may also occur. [1] Myxedema , although included in the name, is not necessarily seen in myxedema coma. [ citation needed ] Coma is also not necessarily seen in myxedema coma. [2] According to newer theories, myxedema coma could result from allostatic overload in a situation where the effects of hypothyroidism are amplified by nonthyroidal illness syndrome . [3] See also [ edit ] Thyroid storm Euthyroid sick syndrome References [ edit ] ^ Berghe, edited by Greet van den; Wartofsky, Leonard (2008). ... You can help Wikipedia by expanding it . v t e v t e Thyroid disease Hypothyroidism Iodine deficiency Cretinism Congenital hypothyroidism Myxedema Myxedema coma Euthyroid sick syndrome Signs and symptoms Queen Anne's sign Woltman sign Thyroid dyshormonogenesis Pickardt syndrome Hyperthyroidism Hyperthyroxinemia Thyroid hormone resistance Familial dysalbuminemic hyperthyroxinemia Hashitoxicosis Thyrotoxicosis factitia Thyroid storm Graves' disease Signs and symptoms Abadie's sign of exophthalmic goiter Boston's sign Dalrymple's sign Stellwag's sign lid lag Griffith's sign Möbius sign Pretibial myxedema Graves' ophthalmopathy Thyroiditis Acute infectious Subacute De Quervain's Subacute lymphocytic Palpation Autoimmune /chronic Hashimoto's Postpartum Riedel's Enlargement Goitre Endemic goitre Toxic nodular goitre Toxic multinodular goiter Thyroid nodule Colloid nodule
AIE causes malabsorption of food, vitamins, and minerals often necessitating replacement fluids and total parenteral nutrition. Some disorders, such as IPEX Syndrome , include autoimmune enteropathy as well as autoimmune "pathies" of the skin, thyroid , other glands , or kidneys . ... Types [ edit ] There are 3 types of autoimmune enteropathy: Type 1 : IPEX syndrome : I mmune dysregulation, P olyendocrinopathy, E nteropathy, X – linked syndrome, which is caused by a mutation in the FOXP3 gene. ... Type 2 : IPEX-like, which manifests similarly to IPEX syndrome but without recognizable mutations in the FOXP3 gene.
Severe-immune mediated enteropathy describes a variety of intestinal disorders that can range from a serious, early-onset systemic disease (IPEX; see this term) to a mild isolated gastrointestinal disease. In children it manifests with severe diarrhea and dehydration in the presence of characteristic antibodies (anti-enterocyte and anti-goblet cell) and in adults with chronic diarrhea, malabsorption and weight loss.
A number sign (#) is used with this entry because of evidence that Warburg-Cinotti syndrome (WRCN) is caused by heterozygous mutation in the DDR2 gene (191311) on chromosome 1q23. Description Warburg-Cinotti syndrome is characterized by progressive corneal neovascularization, keloid formation, chronic skin ulcers, wasting of subcutaneous tissue, flexion contractures of the fingers, and acroosteolysis (Xu et al., 2018). ... Xu et al. (2018) studied 6 patients with Warburg-Cinotti syndrome, including the 2 men previously reported by Warburg et al. (2006) (patient 1) and Cinotti et al. (2013) (patient 2), a mother and 2 affected children (patients 3, 4, and 5), and an unrelated 35-year-old woman (patient 6). ... Molecular Genetics In 6 patients from 4 families with Warburg-Cinotti syndrome, including the Danish man originally reported by Warburg et al. (2006) and the Italian man reported by Cinotti et al. (2013), Xu et al. (2018) identified heterozygosity for missense mutations in the DDR2 gene (L610P, 191311.0006 and Y740C, 191311.0007).
., jet lag , shift work , or other circadian rhythm sleep disorders ); another underlying sleep disorder , such as narcolepsy , sleep apnea , [2] idiopathic hypersomnia , or restless legs syndrome ; disorders such as clinical depression or atypical depression ; tumors, head trauma, anemia, kidney failure , hypothyroidism , or an injury to the central nervous system ; drug abuse; genetic predisposition; vitamin deficiency , such as biotin deficiency ; and particular classes of prescription and over-the-counter medication. ... There is declining usage of other drugs such as methylphenidate (Ritalin), dextroamphetamine (Dexedrine), amphetamine (Adderall), lisdexamfetamine (Vyvanse), methamphetamine (Desoxyn), and pemoline (Cylert), as these psychostimulants may have several adverse effects [5] and may lead to dependency, especially when illicitly misused. See also [ edit ] Kleine-Levin syndrome References [ edit ] ^ Guilleminault, C; Brooks, SN (August 2001). ... External links [ edit ] Classification D ICD - 10 : F51.1 , G47.1 ICD - 9-CM : 291.82 , 292.85 , 307.43 - 307.44 , 327.1 , 780.53 - 780.54 MeSH : D006970 External resources eMedicine : med/3129 v t e Sleep and sleep disorders Stages of sleep cycles Rapid eye movement (REM) Non-rapid eye movement Slow-wave Brain waves Alpha wave Beta wave Delta wave Gamma wave K-complex Mu rhythm PGO waves Sensorimotor rhythm Sleep spindle Theta wave Sleep disorders Dyssomnia Excessive daytime sleepiness Hypersomnia Insomnia Kleine–Levin syndrome Narcolepsy Night eating syndrome Nocturia Sleep apnea Catathrenia Central hypoventilation syndrome Obesity hypoventilation syndrome Obstructive sleep apnea Periodic breathing Sleep state misperception Circadian rhythm disorders Advanced sleep phase disorder Cyclic alternating pattern Delayed sleep phase disorder Irregular sleep–wake rhythm Jet lag Non-24-hour sleep–wake disorder Shift work sleep disorder Parasomnia Bruxism Nightmare disorder Night terror Periodic limb movement disorder Rapid eye movement sleep behavior disorder Sleepwalking Somniloquy Benign phenomena Dreams Exploding head syndrome Hypnic jerk Hypnagogia / Sleep onset Hypnopompic state Sleep paralysis Sleep inertia Somnolence Nocturnal clitoral tumescence Nocturnal penile tumescence Nocturnal emission Treatment Sleep diary Sleep hygiene Sleep induction Hypnosis Lullaby Somnology Polysomnography Other Sleep medicine Behavioral sleep medicine Sleep study Daily life Bed Bunk bed Daybed Four-poster bed Futon Hammock Mattress Sleeping bag Bed bug Bedding Bedroom Bedtime Bedtime story Bedtime toy Biphasic and polyphasic sleep Chronotype Dream diary Microsleep Mouth breathing Nap Nightwear Power nap Second wind Siesta Sleep and creativity Sleep and learning Sleep deprivation / Sleep debt Sleeping while on duty Sleepover Snoring
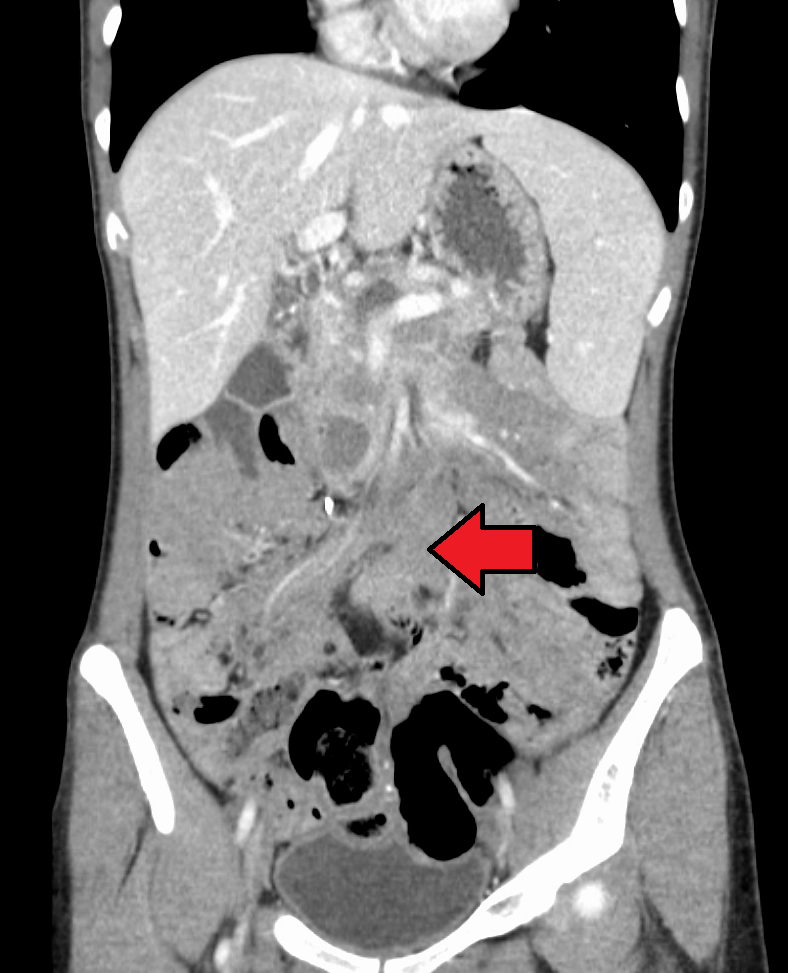
Extranodal nasal NK/T cell lymphoma (NKTCL) is a rare type of cancer. The term extranodal is used because this form of lymphoma is found outside of the traditional lymph node groupings. It mainly affects men around 50 years of age, and usually arises in the nose, paranasal sinuses (paranasal sinuses are cavities (spaces) or small tunnels. located around or near the nose), orbits or upper airway, and that can present with a nasal mass, nasal bleeding, nasal obstruction, a hole in the palate, and mid-facial and/or upper airway destructive lesions. In advanced disease stages, which are associated with a poor prognosis, NKTCL may affect other organs, but usually without enlarged nodes in the body. The treatment depends on the extent of disease and usually involves radiotherapy and chemotherapy.
Extranodal nasal NK/T cell lymphoma (NKTCL) is a rare, malignant neoplasm mainly affecting men in the fifth decade of life, that usually arises in the nose, paranasal sinuses, orbits or upper airway, and that can present with a nasal mass, nasal bleeding, nasal obstruction, palate perforation (i.e. midline perforation of the hard palate), and mid-facial and/or upper airway destructive lesions. In advanced disease stages, which are associated with a poor prognosis, NKTCL may disseminate to other organs. A few cases of NKTCL presenting primarily in the lymph nodes have also been described.
Factor X deficiency is a rare bleeding disorder that varies in severity among affected individuals. The signs and symptoms of this condition can begin at any age, although the most severe cases are apparent in childhood. Factor X deficiency commonly causes nosebleeds, easy bruising, bleeding under the skin, bleeding of the gums, blood in the urine (hematuria), and prolonged or excessive bleeding following surgery or trauma. Women with factor X deficiency can have heavy or prolonged menstrual bleeding (menorrhagia) or excessive bleeding in childbirth, and may be at increased risk of pregnancy loss (miscarriage). Bleeding into joint spaces (hemarthrosis) occasionally occurs. Severely affected individuals have an increased risk of bleeding inside the skull (intracranial hemorrhage), in the lungs (pulmonary hemorrhage), or in the gastrointestinal tract, which can be life-threatening.
A rare inherited bleeding disorder with a decreased antigen and/or activity of factor X (FX) and characterized by mild to severe bleeding symptoms. Epidemiology Prevalence of homozygous forms is estimated at 1/1 000 000. Both sexes are equally affected. Clinical description Congenital FX deficiency manifests at any age but in general, severe forms of the disease manifest early in life. Patients may experience severe umbilical cord stump bleeding, recurrent epistaxis, soft-tissue hemorrhages, menorrhagia, easy bruising, intra cranial hemorrhages, hematuria, hemarthroses and excessive bleeding during or following surgery or delivery or trauma. Etiology Inherited congenital FX deficiency is caused by mutations in the F10 gene (13q34) controlling the production of plasma FX.
Factor X deficiency is a rare disorder that affects the blood's ability to clot. The severity of the disorder and the associated signs and symptoms can vary significantly from person to person. Common features of factor X deficiency may include easy bruising, frequent nosebleeds, bleeding gums, blood in the urine, and prolonged bleeding after minor injuries. Women with factor X deficiency may also experience heavy menstrual bleeding and may have an increased risk for first trimester miscarriages. Acquired (non-inherited) factor X deficiency, which is the most common form of the disorder, generally occurs in people with no family history of the disorder.
Approximately 10% of individuals with Gardner's syndrome , a type of FAP with extracolonic features, have desmoid tumors. [1] Histologically they resemble very low-grade fibrosarcomas , [2] but they are very locally aggressive and tend to recur even after complete resection. ... Even though they occur sporadically, they can also be seen as a part of Gardner's syndrome. A high index of suspicion and a thorough triple examination protocol is necessary to detect rare lesions like a desmoid tumour which can masquerade as breast carcinoma. ... "Desmoid tumour of the breast as a manifestation of Gardner's syndrome" . International Journal of Surgery Case Reports . 3 (5): 139–142. doi : 10.1016/j.ijscr.2012.01.004 . ... External links [ edit ] Classification D ICD - 10 : D48.1 MeSH : D018222 DiseasesDB : 29794 External resources eMedicine : article/1060887 v t e Soft tissue disorders Capsular joint Synoviopathy Synovitis / Tenosynovitis Calcific tendinitis Stenosing tenosynovitis Trigger finger De Quervain syndrome Transient synovitis Ganglion cyst osteochondromatosis Synovial osteochondromatosis Plica syndrome villonodular synovitis Giant-cell tumor of the tendon sheath Bursopathy Bursitis Olecranon Prepatellar Trochanteric Subacromial Achilles Retrocalcaneal Ischial Iliopsoas Synovial cyst Baker's cyst Calcific bursitis Noncapsular joint Symptoms Ligamentous laxity Hypermobility Enthesopathy / Enthesitis / Tendinopathy upper limb Adhesive capsulitis of shoulder Impingement syndrome Rotator cuff tear Golfer's elbow Tennis elbow lower limb Iliotibial band syndrome Patellar tendinitis Achilles tendinitis Calcaneal spur Metatarsalgia Bone spur other/general: Tendinitis / Tendinosis Nonjoint Fasciopathy Fasciitis : Plantar Nodular Necrotizing Eosinophilic Fibromatosis / contracture Dupuytren's contracture Plantar fibromatosis Aggressive fibromatosis Knuckle pads
Intra-abdominal DTs are often observed in patients with an association of familial adenomatous polyposis (FAP) or Gardner syndrome (see these terms). Etiology DTs result from the proliferation of well-differentiated myofibroblasts.
Description Hereditary desmoid disease usually presents as an extraintestinal manifestation of familial adenomatous polyposis (FAP; 175100), also known as Gardner syndrome, which is an autosomal dominant disorder caused by germline mutation in the APC gene. ... Clinical Features Maher et al. (1992) described a mother and son with marked infiltrative fibromatosis of the mesentery of the type that had been observed as part of the Gardner syndrome. However, colonic polyps, osteomas, sebaceous cysts, and congenital hypertrophy of the retinal pigment epithelium (CHRPE) typical of Gardner syndrome were not observed. ... Colonoscopic examination was negative, and other extracolonic features of Gardner syndrome were not found. Family history revealed a maternal history of nonpolyposis colon cancer syndrome and breast tumor.
Radial dysplasia is the condition in which the forearm bone and the soft tissues on the thumb side are underdeveloped or absent. [3] In an embryo the upper extremities develop from week four of the gestation. [1] During the fifth to eighth week the thumb will further develop. [4] In this period something goes wrong with the growth of the thumb but the exact cause of thumb hypoplasia is unknown. [1] One out of every 100,000 live births shows thumb hypoplasia. [2] In more than 50% of the cases both hands are affected, otherwise mainly the right hand is affected. [1] [2] About 86% of the children with hypoplastic thumb have associated abnormalities. [1] [2] Embryological hand development occurs simultaneously with growth and development of the cardiovascular, neurologic and hematopoietic systems. [2] Thumb hypoplasia has been described in 30 syndromes wherein those abnormalities have been seen. A syndrome is a combination of three or more abnormalities. Examples of syndromes with an hypoplastic thumb are Holt-Oram syndrome , VACTERL association [1] and thrombocytopenia absent radius ( TAR syndrome ). [2] Contents 1 Classification 2 Cause 3 Diagnosis 4 Treatment 5 References Classification [ edit ] In general there are five types of thumb hypoplasia, originally described by Muller in 1937 and improved by Blauth, Buck-Gramcko and Manske. [1] - Type I : the thumb is small, normal components are present but undersized. [3] Two muscles of the thumb, the abductor pollicis brevis and opponens pollicis , are not fully developed ,. [2] [3] This type requires no surgical treatment in most cases. [1] [5] - Type II is characterized by a tight web space between the thumb and index finger which restricts movement, [5] poor thenar muscles and an unstable middle joint of the thumb metacarpophalangeal joint . [3] This unstable thumb is best treated with reconstruction of the mentioned structures. [1] - Type III thumbs are subclassified into two subtypes by Manske. ... In this group careful attention should be paid to anomalous tendons coming from the forearm (extrinsic muscles, like an aberrant long thumb flexor – flexor pollicis longus ). [3] [4] [5] - Type IV is called a pouce flottant, floating thumb. [1] [2] [3] [5] This thumb has a neurovascular bundle which connects it to the skin of the hand. [1] [3] [5] There’s no evidence of thenar muscles and rarely functioning tendons. [4] [5] It has a few rudimentary bones. [4] [5] Children with type IV are difficult to reconstruct. [1] [4] This type is nearly always treated with an index finger pollicization to improve hand function. [1] [5] - Type V is no thumb at all [2] [3] and requires pollicization. [1] [5] Type II Type III-A Type III-B Type IV Type V Cause [ edit ] The cause is unknown, and likely related to genetic abnormalities.Children with Fanconi anemia can sometimes display hypoplasia of the thumb. [ citation needed ] Diagnosis [ edit ] Three main points in diagnosing thumb hypoplasia are: width of the first web space, instability of the involved joints and function of the thumb. [5] Thorough physical examination together with anatomic verification at operation reveals all the anomalies. [1] [5] An X-ray of the hand and thumb in two directions is always mandatory. [5] When the pediatrician thinks the condition is associated with some kind of syndrome other tests will be done. [1] More subtle manifestations of types I and II may not be recognized, especially when more obvious manifestations of longitudinal radial deficiency in the opposite extremity are present. ... Congenital Thumb Deformities and Associated Syndromes. Journal of Craniofacial Surgery, vol 20, number 4, 1039–1044 ^ a b c d e f g h i j k l m n o Manske, P.R. & Goldfarb, C.A. (2009).
A number sign (#) is used with this entry because of evidence that Crisponi/cold-induced sweating syndrome-2 (CISS2) is caused by compound heterozygous mutation in the CLCF1 gene (607672) on chromosome 11q13. Description Crisponi/cold-induced sweating syndrome is an autosomal recessive disorder characterized in the neonatal period by orofacial weakness with impaired sucking and swallowing resulting in poor feeding necessitating medical intervention. ... These features, referred to as 'Crisponi syndrome' in infancy, can result in early death without advanced care. ... For a discussion of genetic heterogeneity of Crisponi/cold-induced sweating syndrome, see CISS1 (272430). Clinical Features Rousseau et al. (2006) reported an Australian man, first examined when he was 46 years of age, who stated that he had feeding difficulties as an infant and suffered all his life from profuse sweating on the face, trunk, and upper limbs when cold in addition to inability to sweat in hot weather.
Summary Clinical characteristics. Huppke-Brendel syndrome (HBS) is characterized by bilateral congenital cataracts, sensorineural hearing loss, and severe developmental delay. ... Diagnosis Formal diagnostic criteria for Huppke-Brendel syndrome (HBS) have not been established. ... Clinical Characteristics Clinical Description Huppke-Brendel syndrome (HBS) is characterized by cataract, sensorineural deafness, and severe developmental delay in all reported individuals. ... Disorders to Consider in the Differential Diagnosis of Huppke-Brendel Syndrome View in own window Disorder Gene(s) MOI Overlapping Clinical /Laboratory Features Distinguishing Clinical Features Disorders w/overlapping clinical & laboratory features SUCLG1 -related mtDNA depletion syndrome, encephalomyopathic form w/methylmalonic aciduria SUCLG1 AR Hypotonia Deafness DD Dystonia, other extrapyramidal features, & basal ganglia signal changes on MRI differentiate this disorder from HBS. SUCLA2 -related mtDNA depletion syndrome, encephalomyopathic form w/methylmalonic aciduria SUCLA2 AR Hypotonia Deafness DD Dystonia, extrapyramidal features, & basal ganglia signal changes differentiate this disorder from HBS.
Overview Esophagitis (uh-sof-uh-JIE-tis) is inflammation of the esophagus. The esophagus is the muscular tube that delivers food from your mouth to your stomach. Esophagitis can cause painful, difficult swallowing and chest pain. Many different things can cause esophagitis. Some common causes include stomach acids backing up into the esophagus, infection, oral medicines and allergies. Treatment for esophagitis depends on the underlying cause and how badly the tissue lining the esophagus is damaged.
The EM findings strongly support the diagnosis of ATP6V0A2 -related cutis laxa but are also seen in De Barsy syndrome [Guerra et al 2004] (see Differential Diagnosis). ... Bleeding disorder linked to coagulation factor deficiencies may occur. Wrinkly skin syndrome (WSS) includes many features of Debré-type cutis laxa but is milder [Gazit et al 1973]. ... Pathogenic variants in PYCR1 cause a phenotype which shares many similarities with GO, wrinkly skin syndrome, and De Barsy syndrome. Affected individuals have a common facial gestalt with triangular face, hypomimia, large everted ears, and a cutis laxa more pronounced in extremities. ... ALDH18A1 -related cutis laxa (ARCL3A) (OMIM 219150). A syndrome of IUGR, cataracts, postnatal growth failure and developmental delay with cutis laxa has been described in two pedigrees. Joint hyperlaxity is apparently a common feature. This syndrome falls within de Barsy syndrome spectrum.
Adults may have large, blood-filled bullae in the mouth. [10] If the person's platelet count is between 30,000 and 50,000/mm 3 , bruising with minor trauma may be expected; if it is between 15,000 and 30,000/mm 3 , spontaneous bruising will be seen (mostly on the arms and legs). [11] Causes [ edit ] Thrombocytopenia can be inherited or acquired. [12] Decreased production [ edit ] Abnormally low platelet production may be caused by: [13] Dehydration , Vitamin B 12 or folic acid deficiency Leukemia , myelodysplastic syndrome , or aplastic anemia Decreased production of thrombopoietin by the liver in liver failure Sepsis , systemic viral or bacterial infection Leptospirosis Hereditary syndromes [14] ACTN1-related thrombocytopenia Alport syndrome Amegakaryocytic thrombocytopenia with radio-ulnar synostosis ANKRD26 related thrombocytopenia Autosomal dominant thrombocytopenia Bernard–Soulier syndrome (associated with large platelets) Congenital amegakaryocytic thrombocytopenia Congenital amegakaryocytic thrombocytopenia and radioulnar synostosis CYCS-related thrombocytopenia ETV6 related thrombocytopenia Fanconi anemia Filaminopathies A FYB related thrombocytopenia Glanzmann's thrombasthenia GNE myopathy with congenital thrombocytopenia Gray platelet syndrome Macrothrombocytopenia and hearing loss May–Hegglin anomaly MYH9-related disease PRKACG-related thrombocytopenia Paris-Trousseau thrombocytopenia / Jacobsen syndrome SLFN14-related thrombocytopenia Stormorken syndrome TRPM7-related thrombocytopenia Thrombocytopenia absent radius syndrome Tropomyosin 4-related thrombocytopenia TUBB1-related thrombocytopenia Upshaw–Schulman syndrome Wiskott–Aldrich syndrome X-linked thrombocytopenia X-linked thrombocytopenia with thalassemia Increased destruction [ edit ] TTP Abnormally high rates of platelet destruction may be due to immune or nonimmune conditions, including: [15] Immune thrombocytopenic purpura Thrombotic thrombocytopenic purpura Hemolytic–uremic syndrome Disseminated intravascular coagulation Paroxysmal nocturnal hemoglobinuria Antiphospholipid syndrome Systemic lupus erythematosus Post-transfusion purpura Neonatal alloimmune thrombocytopenia Hypersplenism Dengue fever Gaucher's disease Zika virus Medication-induced [ edit ] These medications can induce thrombocytopenia through direct myelosuppression: [16] Valproic acid Methotrexate Carboplatin Interferon Isotretinoin Panobinostat H 2 blockers and proton-pump inhibitors Other causes [ edit ] Lab error, possibly due to the anticoagulant EDTA in CBC specimen tubes; [ citation needed ] a citrated platelet count is a useful follow-up study [17] Snakebite [18] Niacin toxicity [19] Lyme disease [20] Thrombocytapheresis (also called plateletpheresis) [ citation needed ] Niemann–Pick disease [21] [22] Diagnosis [ edit ] Laboratory tests for thrombocytopenia might include full blood count , liver enzymes , kidney function , vitamin B 12 levels, folic acid levels, erythrocyte sedimentation rate , and peripheral blood smear. ... External links [ edit ] Classification D ICD - 10 : D69.6 ICD - 9-CM : 287.5 MeSH : D013921 External resources Patient UK : Thrombocytopenia Scholia has a topic profile for Thrombocytopenia . v t e Disorders of bleeding and clotting Coagulation · coagulopathy · Bleeding diathesis Clotting By cause Clotting factors Antithrombin III deficiency Protein C deficiency Activated protein C resistance Protein S deficiency Factor V Leiden Prothrombin G20210A Platelets Sticky platelet syndrome Thrombocytosis Essential thrombocythemia DIC Purpura fulminans Antiphospholipid syndrome Clots Thrombophilia Thrombus Thrombosis Virchow's triad Trousseau sign of malignancy By site Deep vein thrombosis Bancroft's sign Homans sign Lisker's sign Louvel's sign Lowenberg's sign Peabody's sign Pratt's sign Rose's sign Pulmonary embolism Renal vein thrombosis Bleeding By cause Thrombocytopenia Thrombocytopenic purpura : ITP Evans syndrome TM TTP Upshaw–Schulman syndrome Heparin-induced thrombocytopenia May–Hegglin anomaly Platelet function adhesion Bernard–Soulier syndrome aggregation Glanzmann's thrombasthenia platelet storage pool deficiency Hermansky–Pudlak syndrome Gray platelet syndrome Clotting factor Hemophilia A/VIII B/IX C/XI von Willebrand disease Hypoprothrombinemia/II Factor VII deficiency Factor X deficiency Factor XII deficiency Factor XIII deficiency Dysfibrinogenemia Congenital afibrinogenemia Signs and symptoms Bleeding Bruise Hematoma Petechia Purpura Nonthrombocytopenic purpura By site head Epistaxis Hemoptysis Intracranial hemorrhage Hyphema Subconjunctival hemorrhage torso Hemothorax Hemopericardium Pulmonary hematoma abdomen Gastrointestinal bleeding Hemobilia Hemoperitoneum Hematocele Hematosalpinx joint Hemarthrosis v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline