Dementia that involves impairments in cognitive function caused by problems in blood vessels that feed the brain Vascular dementia Other names Arteriosclerotic dementia (in the ICD-9) Multi-infarct dementia (in the ICD-10) Vascular cognitive impairment Specialty Psychiatry , neurology Vascular dementia (VaD) is dementia caused by problems in the supply of blood to the brain, typically a series of minor strokes , leading to worsening cognitive decline that occurs step by step. [1] The term refers to a syndrome consisting of a complex interaction of cerebrovascular disease and risk factors that lead to changes in the brain structures due to strokes and lesions , and resulting changes in cognition. The temporal relationship between a stroke and cognitive deficits is needed to make the diagnosis. [2] Contents 1 Signs and symptoms 2 Causes 3 Diagnosis 3.1 Pathology 4 Prevention 5 Treatment 6 Prognosis 7 Epidemiology 8 See also 9 References 10 External links Signs and symptoms [ edit ] Differentiating dementia syndromes can be challenging, due to the frequently overlapping clinical features and related underlying pathology. ... Retrieved December 21, 2007, from https://www.sciencedaily.com/releases/2007/12/071219202948.htm Classification D ICD - 10 : F01.1 ICD - 9-CM : 290.4 MeSH : D015161 DiseasesDB : 8393 External resources MedlinePlus : 000746 eMedicine : med/3150 neuro/227 v t e Mental and behavioral disorders Adult personality and behavior Gender dysphoria Ego-dystonic sexual orientation Paraphilia Fetishism Voyeurism Sexual maturation disorder Sexual relationship disorder Other Factitious disorder Munchausen syndrome Intermittent explosive disorder Dermatillomania Kleptomania Pyromania Trichotillomania Personality disorder Childhood and learning Emotional and behavioral ADHD Conduct disorder ODD Emotional and behavioral disorders Separation anxiety disorder Movement disorders Stereotypic Social functioning DAD RAD Selective mutism Speech Stuttering Cluttering Tic disorder Tourette syndrome Intellectual disability X-linked intellectual disability Lujan–Fryns syndrome Psychological development ( developmental disabilities ) Pervasive Specific Mood (affective) Bipolar Bipolar I Bipolar II Bipolar NOS Cyclothymia Depression Atypical depression Dysthymia Major depressive disorder Melancholic depression Seasonal affective disorder Mania Neurological and symptomatic Autism spectrum Autism Asperger syndrome High-functioning autism PDD-NOS Savant syndrome Dementia AIDS dementia complex Alzheimer's disease Creutzfeldt–Jakob disease Frontotemporal dementia Huntington's disease Mild cognitive impairment Parkinson's disease Pick's disease Sundowning Vascular dementia Wandering Other Delirium Organic brain syndrome Post-concussion syndrome Neurotic , stress -related and somatoform Adjustment Adjustment disorder with depressed mood Anxiety Phobia Agoraphobia Social anxiety Social phobia Anthropophobia Specific social phobia Specific phobia Claustrophobia Other Generalized anxiety disorder OCD Panic attack Panic disorder Stress Acute stress reaction PTSD Dissociative Depersonalization disorder Dissociative identity disorder Fugue state Psychogenic amnesia Somatic symptom Body dysmorphic disorder Conversion disorder Ganser syndrome Globus pharyngis Psychogenic non-epileptic seizures False pregnancy Hypochondriasis Mass psychogenic illness Nosophobia Psychogenic pain Somatization disorder Physiological and physical behavior Eating Anorexia nervosa Bulimia nervosa Rumination syndrome Other specified feeding or eating disorder Nonorganic sleep Hypersomnia Insomnia Parasomnia Night terror Nightmare REM sleep behavior disorder Postnatal Postpartum depression Postpartum psychosis Sexual dysfunction Arousal Erectile dysfunction Female sexual arousal disorder Desire Hypersexuality Hypoactive sexual desire disorder Orgasm Anorgasmia Delayed ejaculation Premature ejaculation Sexual anhedonia Pain Nonorganic dyspareunia Nonorganic vaginismus Psychoactive substances, substance abuse and substance-related Drug overdose Intoxication Physical dependence Rebound effect Stimulant psychosis Substance dependence Withdrawal Schizophrenia , schizotypal and delusional Delusional Delusional disorder Folie à deux Psychosis and schizophrenia-like Brief reactive psychosis Schizoaffective disorder Schizophreniform disorder Schizophrenia Childhood schizophrenia Disorganized (hebephrenic) schizophrenia Paranoid schizophrenia Pseudoneurotic schizophrenia Simple-type schizophrenia Other Catatonia Symptoms and uncategorized Impulse control disorder Klüver–Bucy syndrome Psychomotor agitation Stereotypy v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis
Overview Vascular dementia is a general term describing problems with reasoning, planning, judgment, memory and other thought processes caused by brain damage from impaired blood flow to your brain. You can develop vascular dementia after a stroke blocks an artery in your brain, but strokes don't always cause vascular dementia. Whether a stroke affects your thinking and reasoning depends on your stroke's severity and location. Vascular dementia can also result from other conditions that damage blood vessels and reduce circulation, depriving your brain of vital oxygen and nutrients. Factors that increase your risk of heart disease and stroke — including diabetes, high blood pressure, high cholesterol and smoking — also raise your vascular dementia risk.
Definitions [ edit ] Systemic inflammatory response syndrome [29] Finding Value Temperature <36 °C (96.8 °F) or >38 °C (100.4 °F) Heart rate >90/min Respiratory rate >20/min or PaCO2 <32 mmHg (4.3 kPa) WBC <4x10 9 /L (<4000/mm³), >12x10 9 /L (>12,000/mm³), or ≥10% bands Sepsis Steps. ... On the other hand, systemic inflammatory response syndrome (SIRS) occurs in people without the presence of infection, for example, in those with burns , polytrauma , or the initial state in pancreatitis and chemical pneumonitis . ... Then, an immunosuppression state ensues when the proinflammatory T helper cell 1 (TH1) is shifted to TH2, [47] mediated by interleukin 10 , which is known as "compensatory anti-inflammatory response syndrome". [22] The apoptosis (cell death) of lymphocytes further worsens the immunosuppression. Neutrophils , monocytes , macrophages , dendritic cells , CD4+ T cells , and B cells all undergo apoptosis, whereas regulatory T cells are more apoptosis resistant. [9] Subsequently, multiple organ failure ensues because tissues are unable to use oxygen efficiently due to inhibition of cytochrome c oxidase . [47] Inflammatory responses cause multiple organ dysfunction syndrome through various mechanisms as described below. Increased permeability of the lung vessels causes leaking of fluids into alveoli, which results in pulmonary edema and acute respiratory distress syndrome (ARDS). Impaired utilization of oxygen in the liver impairs bile salt transport, causing jaundice (yellowish discoloration of skin).
Primary sclerosing cholangitis is a condition that affects the bile ducts. These ducts carry bile (a fluid that helps to digest fats) from the liver, where bile is produced, to the gallbladder , where it is stored, and to the small intestine , where it aids in digestion. Primary sclerosing cholangitis occurs because of inflammation in the bile ducts (cholangitis) that leads to scarring (sclerosis) and narrowing of the ducts. As a result, bile cannot be released to the gallbladder and small intestine, and it builds up in the liver . Primary sclerosing cholangitis is usually diagnosed around age 40, and for unknown reasons, it affects men twice as often as women.
Overview Primary sclerosing (skluh-ROHS-ing) cholangitis (koh-lan-JIE-tis) is a disease of the bile ducts. Bile ducts carry the digestive liquid bile from your liver to your small intestine. In primary sclerosing cholangitis, inflammation causes scars within the bile ducts. These scars make the ducts hard and narrow and gradually cause serious liver damage. A majority of people with primary sclerosing cholangitis also have inflammatory bowel disease, such as ulcerative colitis or Crohn's disease.
Clinical Features In a brother and sister, Neuhaus et al. (1997) described progressive tubulointerstitial nephropathy and cholestatic liver disease. The main characteristics were progressive renal failure and elevated liver enzymes. Dialysis was started at the age of approximately 2 years and 6.5 years, respectively. Renal histology showed sclerosing glomeruli and atrophic tubules; the interstitium was fibrotic and infiltrated by lymphocytes. Endoscopic retrograde cholangiopancreatography revealed segmental irregularities and narrowing of the intrahepatic bile ducts, consistent with early primary sclerosing cholangitis.
Primary sclerosing cholangitis (PSC) is a rare, slowly progressive liver disease characterized by inflammation and destruction of the intra- and/or extra-hepatic bile ducts that lead to cholestasis, liver fibrosis, liver cirrhosis and ultimately liver failure. Epidemiology The prevalence of PSC in Europe ranges from 1/446,000-1/6,170. A male preponderance is observed with a male to female ratio of approximately 2:1. Clinical description PSC can occur at any age with a peak incidence around 40 years of age. Symptoms and clinical findings are variable, depending on the stage of the disease.
Primary sclerosing cholangitis (PSC) is characterized by inflammation in the bile ducts (cholangitis) that leads to scarring (sclerosis), narrowing of the ducts, and a buildup of bile in the liver. Early signs and symptoms include extreme tiredness, abdominal pain, and itchiness. As the condition worsens it may cause jaundice, an enlarged spleen, and eventually liver cirrhosis and failure. Other complications may include weight loss, vitamin deficiency, and osteoporosis. Many people with PSC develop other autoimmune conditions such as inflammatory bowel disease , type 1 diabetes , celiac disease , or thyroid disease .
Description Primary sclerosing cholangitis (PSC) is a slowly progressive cholestatic liver disease characterized by fibroobliterative inflammation of the biliary tract, leading to cirrhosis and portal hypertension. It is a major indication for liver transplantation (Sheth et al., 2003). Approximately 75 to 80% of PSC cases are associated with inflammatory bowel disease (IBD; see 266600), and 2.5 to 7.5% of patients with IBD develop PSC (Lee and Kaplan, 1995). Mapping In a genomewide association study of in 715 Scandinavian and German individuals with PSC and 2,962 controls, Melum et al. (2011) found the strongest associations with SNPs in the HLA complex at chromosome 6p21, peaking at rs3134792 in HLA-B (142830) (p = 6.8 x 10(-49)). Carriers of the G allele at rs3134792 were HLA-B*08 carriers in 99% of the cases, and were HLA-DRB1*03 (142857) carriers in 90% of the cases.
However, the workgroup also concluded that it was unlikely that MCS would receive extensive financial resources from federal agencies because of budgetary constraints and the allocation of funds to other, extensively overlapping syndromes with unknown cause , such as chronic fatigue syndrome, fibromyalgia, and Gulf War syndrome. ... "Panic response to sodium lactate infusion in patients with multiple chemical sensitivity syndrome". J Allergy Clin Immunol . 99 (4): 570–4. doi : 10.1016/s0091-6749(97)70086-1 . ... PMID 15165662 . ^ a b Black DW, Doebbeling BN, Voelker MD, Clarke WR, Woolson RF, Barrett DH, Schwartz DA (April 2000). "Multiple chemical sensitivity syndrome: symptom prevalence and risk factors in a military population" . ... Multi-symptom illnesses, unexplained illness, and Gulf War Syndrome " " (PDF) . Psychological Medicine . 36 (6): 735–747. doi : 10.1017/s0033291705006975 . ... "Multi-symptom illnesses, unexplained illness, and Gulf War Syndrome" . Philosophical Transactions of the Royal Society B . 361 (1468): 543–551. doi : 10.1098/rstb.2006.1815 .
It can be defined as an excess of amino acid and protein metabolism end products, such as urea and creatinine , in the blood that would be normally excreted in the urine. Uremic syndrome can be defined as the terminal clinical manifestation of kidney failure (also called renal failure ). [1] It is the signs, symptoms and results from laboratory tests which result from inadequate excretory, regulatory, and endocrine function of the kidneys. [2] Both uremia and uremic syndrome have been used interchangeably to denote a very high plasma urea concentration that is the result of renal failure. [1] The former denotation will be used for the rest of the article. ... Treatment can be by dialysis or a kidney transplant , though some patients choose to pursue symptom control and conservative care instead. [3] Contents 1 Signs and symptoms 1.1 Residual syndrome 2 Causes 3 Diagnosis 3.1 Blood tests 3.2 Urine tests 3.3 Radioisotope tests 3.4 Other 4 Mechanism 4.1 Uremic toxins 4.2 Biochemical characteristics 5 History 6 Oral manifestations 7 Dental considerations 8 Notes 9 References 10 External links Signs and symptoms [ edit ] Classical signs of uremia are: progressive weakness and easy fatigue, loss of appetite due to nausea and vomiting, muscle atrophy , tremors, abnormal mental function, frequent shallow respiration, and metabolic acidosis . ... GFR and their effects [3] GFR (mL/min) Effects 100–120 Normal GFR <60 Uremic symptoms may be present, reduced well-being 30–60 Cognitive impairment 55 Fatigue and reduced stamina <50 Insulin resistance <30 Increasing likelihood of symptoms ≤15 Kidney failure Residual syndrome [ edit ] People on dialysis acquire what is known as "residual syndrome". [5] Residual syndrome is a non-life-threatening disease which is displayed as toxic effects causing many of the same signs and symptoms that uremia displays. ... Frerich described clinical uremic syndrome and suggested that a toxicity was the mechanism of its cause. ... External links [ edit ] Uremia , WebMD.com Understanding Uremia , uspharmacist.com Classification D ICD - 10 : N19 , R39.2 ICD - 9-CM : 585 - 586 , 788.9 MeSH : D014511 DiseasesDB : 26060 External resources eMedicine : med/2341 v t e Kidney disease Glomerular disease See Template:Glomerular disease Tubules Renal tubular acidosis proximal distal Acute tubular necrosis Genetic Fanconi syndrome Bartter syndrome Gitelman syndrome Liddle's syndrome Interstitium Interstitial nephritis Pyelonephritis Balkan endemic nephropathy Vascular Renal artery stenosis Renal ischemia Hypertensive nephropathy Renovascular hypertension Renal cortical necrosis General syndromes Nephritis Nephrosis Renal failure Acute renal failure Chronic kidney disease Uremia Other Analgesic nephropathy Renal osteodystrophy Nephroptosis Abderhalden–Kaufmann–Lignac syndrome Diabetes insipidus Nephrogenic Renal papilla Renal papillary necrosis Major calyx / pelvis Hydronephrosis Pyonephrosis Reflux nephropathy v t e Symptoms and signs relating to the urinary system Pain Dysuria Renal colic Costovertebral angle tenderness Vesical tenesmus Control Urinary incontinence Enuresis Diurnal enuresis Giggling Nocturnal enuresis Post-void dribbling Stress Urge Overflow Urinary retention Volume Oliguria Anuria Polyuria Other Lower urinary tract symptoms Nocturia urgency frequency Extravasation of urine Uremia Eponymous Addis count Brewer infarcts Lloyd's sign Mathe's sign Authority control NDL : 00575462
In individuals with thoracic insufficiency syndrome, avoid supplemental oxygen, which can suppress respiratory drive. ... Limb reduction defects that may affect the fingers in atypical or nonclassic FOP and may be mistaken for a brachydactyly syndrome in individuals who have not yet developed heterotopic ossifications Imaging findings (see Figure 1) Figure 1. ... Scoliosis affects up to 65% of individuals, may be rapidly progressive due to paravertebral lesions, and may contribute to thoracic insufficiency syndrome. Pelvic radiographs may identify congenital short broad femoral necks , which rarely affect function. ... Mechanical respiratory difficulties incl thoracic insufficiency syndrome Singing, swimming, incentive spirometry Positive pressure ventilation when indicated Avoid respiratory infections. ... In individuals with thoracic insufficiency syndrome, avoid supplemental oxygen, which can suppress respiratory drive.
Fibrodysplasia ossificans progressiva is a disorder in which muscle tissue and connective tissue such as tendons and ligaments are gradually replaced by bone (ossified), forming bone outside the skeleton (extra-skeletal or heterotopic bone) that constrains movement. This process generally becomes noticeable in early childhood, starting with the neck and shoulders and proceeding down the body and into the limbs. Extra-skeletal bone formation causes progressive loss of mobility as the joints become affected. Inability to fully open the mouth may cause difficulty in speaking and eating. Over time, people with this disorder may experience malnutrition due to their eating problems.
Fibrodysplasia ossificans progressiva (FOP) is a disorder in which skeletal muscle and connective tissue , such as tendons and ligaments, are gradually replaced by bone (ossified). This condition leads to bone formation outside the skeleton (extra-skeletal or heterotopic bone) that restricts movement. This process generally becomes noticeable in early childhood, starting with the neck and shoulders and moving down the body and into the limbs. People with FOP are born with abnormal big toes (hallux valgus) which can be helpful in making the diagnosis. Trauma, such as a fall or invasive medical procedure, or a viral illness may trigger episodes of muscle swelling and inflammation (myositis).
A number sign (#) is used with this entry because fibrodysplasia ossificans progressiva (FOP) is caused by heterozygous mutation in the ACVR1 gene (102576) on chromosome 2q24. Description Fibrodysplasia ossificans progressiva is a rare autosomal dominant disease with complete penetrance involving progressive ossification of skeletal muscle, fascia, tendons, and ligaments. FOP has a prevalence of approximately 1 in 2 million worldwide, and shows no geographic, ethnic, racial, or gender preference. Individuals with FOP appear normal at birth except for great toe abnormalities: the great toes are short, deviated, and monophalangic. Ossification occurs progressively over the course of a lifetime in an inevitable and unpredictable episodic manner, with most patients being confined to a wheelchair by the third decade of life and requiring lifelong care (summary by Petrie et al., 2009).
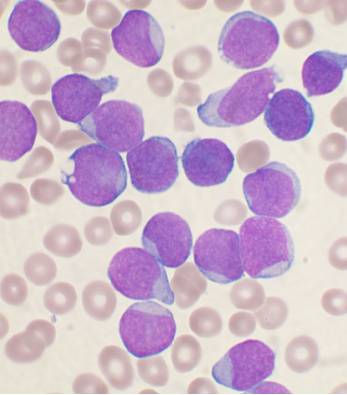
Specialty Hematology , oncology Symptoms Feeling tired, pale color, fever, easy bleeding or bruising, bone pain, enlarged lymph nodes [1] Complications Infection , tumor lysis syndrome [2] [3] Usual onset 2–5 years old [4] Types B-cell ALL , T-cell ALL [2] Causes Usually unknown [5] Risk factors Identical twin with ALL, Down syndrome , Fanconi anemia , ataxia telangiectasia , Klinefelter syndrome , high birth weight , significant radiation exposure [6] [5] [1] Diagnostic method Blood tests and bone marrow examination [3] Differential diagnosis Infectious mononucleosis , acute myeloid leukemia , lymphoblastic lymphoma , aplastic anemia [3] Treatment Chemotherapy , stem cell transplantation , radiation therapy , targeted therapy [1] Prognosis Children : 90% five-year survival rate [2] Adults : 35% five-year survival [7] Frequency 1 in 1,750 children [4] [8] Deaths 111,000 (2015) [9] Acute lymphoblastic leukemia ( ALL ) is a cancer of the lymphoid line of blood cells characterized by the development of large numbers of immature lymphocytes . [1] Symptoms may include feeling tired, pale skin color, fever , easy bleeding or bruising, enlarged lymph nodes , or bone pain. [1] As an acute leukemia , ALL progresses rapidly and is typically fatal within weeks or months if left untreated. [10] In most cases, the cause is unknown. [2] Genetic risk factors may include Down syndrome , Li-Fraumeni syndrome , or neurofibromatosis type 1 . [1] Environmental risk factors may include significant radiation exposure or prior chemotherapy . [1] Evidence regarding electromagnetic fields or pesticides is unclear. [4] [6] Some hypothesize that an abnormal immune response to a common infection may be a trigger. [4] The underlying mechanism involves multiple genetic mutations that results in rapid cell division . [2] The excessive immature lymphocytes in the bone marrow interfere with the production of new red blood cells , white blood cells , and platelets . [1] Diagnosis is typically based on blood tests and bone marrow examination . [3] ALL is typically treated initially with chemotherapy aimed at bringing about remission . [2] This is then followed by further chemotherapy typically over a number of years. [2] Additional treatments may include intrathecal chemotherapy or radiation therapy if spread to the brain has occurred. [2] Stem cell transplantation may be used if the disease recurs following standard treatment. [2] Additional treatments such as Chimeric antigen receptor T cell immunotherapy are being used and further studied. [2] ALL affected about 876,000 people globally in 2015 and resulted in about 111,000 deaths. [11] [9] It occurs most commonly in children, particularly those between the ages of two and five. [12] [4] In the United States it is the most common cause of cancer and death from cancer among children. [2] ALL is notable for being the first disseminated cancer to be cured. [13] Survival for children increased from under 10% in the 1960s to 90% in 2015. [2] Survival rates remain lower for babies (50%) [14] and adults (35%). [7] Contents 1 Signs and symptoms 2 Cause 2.1 Risk factors 2.1.1 Genetics 2.1.2 Environmental 2.1.2.1 Infections 3 Mechanism 4 Diagnosis 4.1 Immunophenotyping 4.2 Cytogenetics 4.3 Classification 5 Treatment 5.1 Chemotherapy 5.2 Radiation therapy 5.3 Biological therapy 5.4 Immunotherapy 5.5 Relapsed ALL 5.6 Side effects 5.7 Supportive therapy 6 Prognosis 7 Epidemiology 8 Pregnancy 9 References 10 External links Signs and symptoms [ edit ] Initial symptoms can be nonspecific, particularly in children. ... These genes, in turn, increase the risk that more mutations will occur in developing lymphoid cells. Certain genetic syndromes, like Down Syndrome , have the same effect. ... For instance, the ARID5B mutation is less common in ethnic African populations. [4] Several genetic syndrome also carry increased risk of ALL. These include: Down syndrome , Fanconi anemia , Bloom syndrome , X-linked agammaglobulinemia , severe combined immunodeficiency , Shwachman-Diamond syndrome , Kostmann syndrome , neurofibromatosis type 1 , ataxia-telangiectasia , paroxysmal nocturnal hemoglobinuria , and Li-Fraumeni syndrome . [13] Fewer than 5% of cases are associated with a known genetic syndrome. [7] Rare mutations in ETV6 and PAX5 are associated with a familial form of ALL with autosomal dominant patterns of inheritance . [2] Environmental [ edit ] The environmental exposures that contribute to emergence of ALL is contentious and a subject of ongoing debate. [6] [4] High levels of radiation exposure from nuclear fallout is a known risk factor for developing leukemia. [23] Evidence whether lesser radiation, as from x-ray imaging during pregnancy, increases risk of disease remains inconclusive. [6] Studies that have identified an association between x-ray imaging during pregnancy and ALL found only a slightly increased risk. [4] Exposure to strong electromagnetic radiation from power lines has also been associated with a slightly increased risk of ALL. ... "Secondary leukemia or myelodysplastic syndrome after treatment with epipodophyllotoxins".
For a discussion of genetic heterogeneity of susceptibility to acute lymphoblastic leukemia, see ALL1 (613065). Mapping In a genomewide association study of 317 patient with ALL and 17,958 controls, Trevino et al. (2009) found an association between ALL and 3 SNPs on chromosome 7p12.2. These were in linkage disequilibrium and located in the IKZF1 gene (603023) (rs11978267, p = 8.8 x 10(-11)) and the DDC gene (107930) (rs2167364, p = 2.8 x 10(-6) and rs2242041, p = 9.9 x 10(-7)). Trevino et al. (2009) concluded that germline variants can affect susceptibility to, and characteristics of, ALL and specific ALL subtypes. In a genomewide association study of 2 case-control series, totaling 907 ALL cases and 2,398 controls, Papaemmanuil et al. (2009) found a significant association between ALL and a SNP at chromosome 7p12.2 in the IKZF1 gene (rs4132601, OR of 1.69, p = 1.20 x 10(-19)).
Patients with acute lymphoblastic leukemia (ALL) who present with bulky disease of the lymph nodes, spleen, and mediastinum, so-called lymphomatous ALL (LALL), appear clinically to represent a distinct category of ALL of T-cell lineage. The biologic basis of this distinction was pointed out by Chilcote et al. (1985) who found that 6 of 8 patients with clinical features of LALL had karyotypic abnormalities leading to loss of bands 9p22-p21. The mechanisms varied and included deletions, unbalanced translocations, and loss of the entire chromosome. Only 1 of 57 patients without LALL had an abnormality of chromosome 9 at diagnosis. Loss of a 'suppressor' gene (RB1; 614041) comparable to that in retinoblastoma (180200) was postulated.
A rare disease characterized by malignant proliferation of lymphoid cells blocked at an early stage of differentiation and accounts for 75% of all cases of childhood leukaemia. Epidemiology About 3,000 children in the United States and 5,000 children in Europe are diagnosed with ALL per year. Clinical description The peak incidence occurs between 2 and 5 years of age. ALL may be either asymptomatic or acute with a life-threatening haemorrhage, infection, or episode of respiratory distress. Although ALL primarily affects the bone marrow and peripheral blood, any organ or tissue may be infiltrated by the abnormal cells.
The risk to develop ALL may also be increased by past treatment for cancer, and by having certain genetic conditions or syndromes. Having one or more risk factors does not mean that a person definitely will develop ALL.
They identified the P2RY8/CRLF2 fusion in 7% of individuals with B-progenitor ALL and 53% of individuals with ALL associated with Down syndrome. CRLF2 alteration was associated with activating JAK mutations, and expression of human P2RY8/CRLF2 together with mutated mouse Jak2 (147796) resulted in constitutive JAK-STAT activation and cytokine-independent growth of Ba/F3 cells overexpressing IL7 receptor-alpha (IL7R; 146661).
Disease caused by severe acute respiratory syndrome coronavirus 2 "COVID" and "COVID-19" redirect here. ... Coronavirus disease 2019 (COVID-19) Other names The coronavirus 2019-nCoV acute respiratory disease Novel coronavirus pneumonia [1] [2] Severe pneumonia with novel pathogens [3] False-color transmission electron microscope image of coronavirus Pronunciation / k ə ˈ r oʊ n ə ˌ v aɪ r ə s d ɪ ˈ z iː z / / ˌ k oʊ v ɪ d n aɪ n ˈ t iː n , ˌ k ɒ v ɪ d -/ [4] Specialty Infectious disease Symptoms Fever, cough, fatigue, shortness of breath, loss of taste or smell; sometimes no symptoms at all [5] [6] Complications Pneumonia , viral sepsis , acute respiratory distress syndrome , kidney failure , cytokine release syndrome , respiratory failure , pulmonary fibrosis , pediatric multisystem inflammatory syndrome , chronic COVID syndrome Usual onset 2–14 days (typically 5) from infection Duration 5 days to 10+ months known Causes Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Diagnostic method rRT-PCR testing , CT scan Prevention Hand washing , face coverings, quarantine , physical/ social distancing [7] Treatment Symptomatic and supportive Frequency 95,179,173 [8] confirmed cases Deaths 2,033,641 [8] Part of a series on the COVID-19 pandemic SARS-CoV-2 (virus) COVID-19 (disease) Timeline 2019 2020 January February responses March responses April responses May responses June responses July responses August responses September responses October responses November responses December responses 2021 January responses Locations By continent Africa Antarctica Asia Europe North America Oceania South America By conveyance Cruise ships Naval ships Cases Deaths International response United Nations National responses India New Zealand Philippines Russia Sweden U.K. ... Tourism Video games COVID-19 Portal v t e Coronavirus disease 2019 ( COVID-19 ) is a contagious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The first case was identified in Wuhan , China , in December 2019. ... Around one in five infected individuals do not develop any symptoms. [9] While most people have mild symptoms, some people develop acute respiratory distress syndrome (ARDS). ARDS can be precipitated by cytokine storms , [10] multi-organ failure , septic shock , and blood clots . ... Virology Main article: Severe acute respiratory syndrome coronavirus 2 See also: Variants of SARS-CoV-2 Illustration of SARSr-CoV virion Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel severe acute respiratory syndrome coronavirus.
Department of Veterans Affairs . [4] Before 1998, the terms Gulf War syndrome, Gulf War veterans' illness, unexplained illness , and undiagnosed illness were used interchangeably to describe chronic unexplained symptoms in veterans of the 1991 Gulf War. ... An amino acid supplement containing L-carnosine was found to reduce irritable bowel syndrome-associated diarrhea in a randomized, controlled GWIRP-funded trial in GW Veterans (Baraniuk, 2013). ... PMID 17707114 . ^ Gronseth GS (May 2005). "Gulf war syndrome: a toxic exposure? A systematic review". Neurol Clin . 23 (2): 523–540. doi : 10.1016/j.ncl.2004.12.011 . PMID 15757795 . ^ "Gulf War Syndrome" . University of Virginia . Archived from the original on 14 July 2004. ^ a b c d e Stencel, C (9 April 2010). ... ISBN 978-0-8330-2680-4 . ^ "Campaigners hail 'nerve gas link' to Gulf War Syndrome" . The Scotsman . Edinburgh, UK. 13 November 2004.
AP has been reported anecdotally in the setting of certain neurologic disorders, particularly autism and Williams syndrome (Sacks, 1995). A proportion of individuals with Williams syndrome (194050) are gifted musically, even in the face of significant mental disability. Lenhoff et al. (2001) evaluated 5 patients with Williams syndrome for absolute musical pitch. The 5 patients had a mean IQ of 58 but were able to read musical notation. ... Lenhoff et al. (2001) suggested that the prevalence of AP in individuals with Williams syndrome is higher than that in the general Western population (1 in 10,000), and noted that the age window of AP acquisition in Williams syndrome appears to be extended compared to the general population. The findings are important in the study of the genetics of pitch ability since individuals with Williams syndrome are genetically homogeneous. Hickok et al. (1995) reported that brain imaging of patients with Williams syndrome suggested an exaggerated left-right asymmetry of the planum temporale, which had also been found in musicians with absolute pitch (Schlaug et al., 1995), suggesting a neuroanatomic correlate to the ability.
A number sign (#) is used with this entry because vasculitis, autoinflammation, immunodeficiency, and hematologic defects syndrome (VAIHS) is caused by homozygous or compound heterozygous mutation in the CECR1 gene (ADA2; 607575) on chromosome 22q11. Mutation in the ADA2 gene can also cause Sneddon syndrome (182410), which shows later onset. Description Vasculitis, autoinflammation, immunodeficiency, and hematologic defects syndrome (VAIHS) is an autosomal recessive multisystem disorder with onset in childhood. ... INHERITANCE - Autosomal recessive HEAD & NECK Face - Facial nerve palsies Eyes - Ophthalmoplegia - Optic atrophy (1 patient) CARDIOVASCULAR Vascular - Vasculitis, small and medium vessels - Stroke, ischemic - Stroke, hemorrhagic - Polyarteritis nodosa - Aneurysms - Stenosis - Hypertension (in some patients) ABDOMEN Liver - Hepatomegaly Spleen - Splenomegaly Gastrointestinal - Gastrointestinal pain GENITOURINARY Kidneys - Renal artery aneurysms SKELETAL - Arthritis Hands - Ischemic digital necrosis Feet - Ischemic digital necrosis SKIN, NAILS, & HAIR Skin - Livedo racemosa - Livedo reticularis - Erythema nodosum - Urticarial rash - Purpura Skin Histology - Vasculitis in the reticular dermis - Inflammatory infiltrate - Interstitial neutrophils and macrophages - Perivascular T lymphocytes - Leukocytoclastic vasculitis - Panniculitis MUSCLE, SOFT TISSUES - Myalgia NEUROLOGIC Central Nervous System - Neurologic sequelae of stroke - Altered mental status - Hemiplegia - Headache - Ataxia - Agitation - Cranial nerve dysfunction - Aphasia - Lacunar infarcts in the deep-brain nuclei, brainstem, internal capsule seen on imaging Peripheral Nervous System - Raynaud phenomenon - Neuropathy METABOLIC FEATURES - Fever, recurrent HEMATOLOGY - Lupus anticoagulant (in some patients) - Anemia (in some patients) - Thrombocytosis (in some patients) IMMUNOLOGY - Immunodeficiency - Hypogammaglobulinemia (in some patients) - Leukopenia - Leukocytosis LABORATORY ABNORMALITIES - Abnormal liver enzymes - Acute-phase reactants during fever MISCELLANEOUS - Variable age at onset, usually in first decade, but can occur later - Variable manifestations - Variable severity MOLECULAR BASIS - Caused by mutation in the cat eye syndrome chromosome region, candidate 1 gene (CECR1, 607575.0001 ) ▲ Close
Vasculitis due to ADA2 deficiency is a rare, genetic, systemic and rheumatologic disease due to adenosine deaminase-2 inactivating mutations, combining variable features of autoinflammation, vasculitis, and a mild immunodeficiency. Variable clinical presentation includes chronic or recurrent systemic inflammation with fever, livedo reticularis or racemosa, early-onset ischemic or hemorrhagic strokes, peripheral neuropathy, abdominal pain, hepatosplenomegaly, portal hypertension, cutaneous polyarteritis nodosa, variable cytopenia and immunoglobulin deficiency.
Adenosine Deaminase 2 deficiency (ADA2 deficiency) causes swelling of the blood vessels (vasculitis), leading to decreased blood flow affecting the organs and skin. Though the severity and age of onset can vary, most people with ADA2 deficiency begin having symptoms in childhood. Symptoms may include the following: recurrent strokes at an early age, fevers, pain, an enlarged liver or spleen, and areas of skin discoloration known as livedo racemosa or livedo reticularis . The strokes can affect physical or cognitive functioning. Other symptoms may include arthritis, immune system abnormalities, anemia, and damage to the nervous system. ADA2 deficiency is thought to be caused by genetic changes in the CECR1 gene and inherited in an autosomal recessive pattern.
A group of acute febrile tick-borne diseases characterized by an overlapping clinical picture that includes fever, headache, myalgias, arthralgias, skin eruptions, gastrointestinal symptoms and neurological manifestations. Diseases in this group include human monocytotropic ehrlichiosis (HME), human granulocytotropic anaplasmosis (HGA), and human ehrlichiosis ewingii (HEE).
Osteomalacia is a disease that is characterized by a weakening of the bone, often due to a deficiency of vitamin D . This vitamin supports the development of the bones of the body, so when there are low levels of vitamin D, the bones are not strong enough. Symptoms of osteomalacia can include muscle weakness, bone pain, and walking with a waddling gait. Pain is especially likely to occur in the lower back, hips, and legs. The weakening of the bones may also cause them to easily fracture. Osetomalacia can be caused by having a low level of vitamin D in the diet or lack of sun exposure.
OFG includes granulomatous cheilitis (when it presents as a persistent or recurrent lip swelling), and Melkersson-Rosenthal syndrome (which includes CG, facial nerve palsy and fissured tongue) that can manifest with only CG.