Of note, some individuals with a CHD2 pathogenic variant have a diagnosis of eyelid myoclonia with absences (EMA), a particularly photosensitive epilepsy syndrome [Galizia et al 2015]. Seizures are generally refractory to currently available antiepileptic drugs (AEDs). ... Differential Diagnosis Myoclonic absence epilepsy (MAE) (also known as Doose syndrome) was the original diagnosis in three of six individuals with a de novo CHD2 pathogenic variant [Carvill et al 2013]. ... See STXBP1 Encephalopathy with Epilepsy. Dravet syndrome , an infantile epileptic encephalopathy with onset before age 15 months, is characterized by hemiclonic or generalized seizures that are often triggered and prolonged by fever. ... Over 75% of individuals with Dravet syndrome have a de novo SCN1A pathogenic variant [Marini et al 2011]. In contrast to the seizures of Dravet syndrome, the seizures of CHD2 -related neurodevelopmental disorders are not commonly triggered by fever (4/15 cases) [Suls et al 2013, Thomas et al 2015, Trivisano et al 2015].
The disorder is usually evident at birth, with short stature and multiple joint dislocations or subluxations that dominate the neonatal clinical and radiographic picture, and affected individuals may receive an initial clinical diagnosis of Larsen syndrome (see 245600) or humerospinal dysostosis. ... Hermanns et al. (2008) identified homozygosity or compound heterozygosity for 9 different mutations in the CHST3 gene (see, e.g., 603799.0002-603799.0006) in 6 unrelated patients born with joint dislocations, including 3 patients who carried a diagnosis of recessive Larsen syndrome (245600) and 3 who had been diagnosed with humerospinal dysostosis (HSD); 1 of the latter patients (see 603799.0005) was the boy originally reported by Cortina et al. (1979). None of the patients had the typical flattened facies of Larsen syndrome. Hermanns et al. (2008) stated that the single feature most useful in recognizing CHST3 deficiency seemed to be dysostotic changes in the thoracolumbar spine, namely, widening of the interpedicular distance at L1 on anteroposterior projection and the short and cleft vertebral bodies on lateral projection, which was seen in their patients and was mentioned by both Kozlowski et al. (1974) and Cortina et al. (1979) in their descriptions of HSD; Hermanns et al. (2008) noted that this radiologic feature was also recognizable in Figure 1 of the report on Omani-type SED by Rajab et al. (2004). Given the relatively narrow phenotypic spectrum of these conditions, Hermanns et al. (2008) suggested that the disorders previously designated as Omani-type spondyloepiphyseal dysplasia and humerospinal dysostosis, as well as some patients given a diagnosis of recessive Larsen syndrome, might represent different age-related descriptions of the same condition. ... The patients had presented with various diagnoses, including 15 who had been diagnosed with Larsen syndrome (see, e.g., 603799.0011 and 603799.0012), 2 with chondrodysplasia with multiple dislocations (previously reported by Megarbane and Ghanem, 2004 and Megarbane, 2007; see, e.g., 603799.0002), 1 with humerospinal dysostosis (originally reported by Hall, 1997; see 603799.0010), 1 with Desbuquois syndrome (see 251450), and 1 with spondyloepiphyseal dysplasia.
CHST3-related skeletal dysplasia is a very rare bone disorder characterized clinically by short stature of prenatal onset; dislocation of the knees, hips or elbows; club feet; limitation of range of motion of large joints; progressive kyphosis; and occasional scoliosis. In a few patients, minor heart valve dysplasia has also been described. Intellect, vision and hearing are normal.
Adult T-cell leukemia/lymphoma (ATL) is is a rare and aggressive T-cell lymphoma that is linked to infection by the human T-cell lymphotropic virus 1 (HTLV-1) . The exact mechanism by which HTLV-I infection causes the ATL is unknown. The clinical features of ATL include generalized swelling of the lymph nodes ( lymphadenopathy ), increased liver and spleen size (hepatosplenomegaly), immunosuppression, high levels of calcium in the blood, lytic bone lesions (spots that appear as “holes” on a standard bone x-ray), and skin lesions. There are four basic clinical variants of ATL: acute (60% of cases), lymphomatous (20 % of cases), chronic (10% of cases) and smoldering (10% of cases). The best treatment for these patients is unclear and patients should be enrolled in clinical trials whenever possible.
A rare, virus associated tumor due to human T-cell leukemia virus type 1 or human T-cell lymphotropic virus type 1 (HTLV-1) and is characterized by the presence of anti-HTLV-1 antibodies, and malignant, mature, medium-sized T cells with condensed chromatin and polylobated nuclei. The malignant cells exhibit a mature CD4+ T cells phenotype and express CD2, CD5, CD25, CD45RO, HLA-DR, and T-cell receptor αβ. Presentation is heterogeneous and is typically of aggressive leukemia or lymphoma, variable skin eruptions, and visceral organ involvement.
The exact underlying cause of primary CNS lymphoma is poorly understood; however, people with a weakened immune system (such as those with acquired immunodeficiency syndrome ) or who have had an organ transplant appear to have an increased risk of developing the condition.
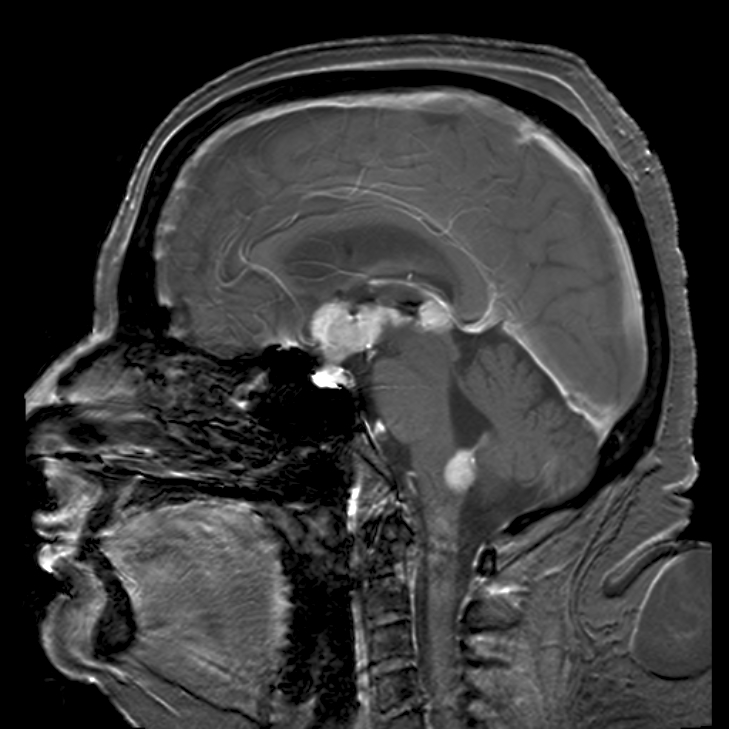
Primary central nervous system lymphoma (PCNSL) is a rare nervous system tumor, predominantly due to diffuse large B-cell lymphoma, that involves brain, leptomeninges, eyes, or rarely spinal cord, in the absence of systemic diffusion at the time of diagnosis. It is characterized by a solitary tumor that, depending on its location, can lead to a variety of symptoms such as headache, nausea, vomiting (and other signs of raised intracranial pressure), focal neurologic deficits, neuropsychiatric and ocular symptoms, seizures and personality changes.
Soon after birth, she had respiratory insufficiency and increased serum lactate, and she later showed hypotonia, failure to thrive, global developmental delay, and Wolff-Parkinson-White syndrome. In the first 2 years of life, she experienced acute deterioration during infections, but her condition later stabilized, and she demonstrated catch-up growth and learned to walk and talk at 3.5 years of age. ... INHERITANCE - Autosomal recessive GROWTH Other - Failure to thrive - Poor overall growth HEAD & NECK Face - Dysmorphic facial features, mild - Small midface Ears - Low-set ears - Retroverted ears Eyes - Hypertelorism Nose - Flat nasal bridge CARDIOVASCULAR Heart - Hypertrophic cardiomyopathy - Impaired left ventricular contractility - Wolff-Parkinson-White syndrome RESPIRATORY - Respiratory insufficiency MUSCLE, SOFT TISSUES - Hypotonia Skeletal muscle shows decreased activity of mitochondrial complex IV - Abnormal mitochondrial morphology NEUROLOGIC Central Nervous System - Global developmental delay - Delayed walking - Impaired intellectual development - Speech delay METABOLIC FEATURES - Lactic acidosis LABORATORY ABNORMALITIES - Combined oxidative phosphorylation deficiency in various tissues - Increased serum lactate MISCELLANEOUS - Prenatal onset - One patient has been reported (last curated April 2019) MOLECULAR BASIS - Caused by mutation in the mitochondrial ribosomal protein S14 gene (MRPS14, 611978.0001 ) ▲ Close
A rare form of hereditary spastic paraplegia which usually presents in late adolescence or early adulthood as a pure phenotype of lower limb spasticity with hyperreflexia and extensor plantar responses, as well as mild bladder disturbances and pes cavus. Rarely, it can present as a complex phenotype with additional manifestations including epilepsy, variable peripheral neuropathy and/or memory impairment.
Familial ventricular tachycardia is usually attributable to recognized conditions such as arrhythmogenic right ventricular dysplasia, hypertrophic cardiomyopathy, familial cardiomyopathy, or one of the long QT syndromes. There are families with ventricular tachycardia in which no recognized underlying condition has been identified. ... Idiopathic ventricular tachycardia is a generic term that describes the various forms of ventricular arrhythmias that occur in patients without structural heart disease and in the absence of long QT syndrome. Many of these tachycardias are focal in origin, localize to the right ventricular outflow tract (RVOT), terminate in response to beta-blockers, verapamil, vagal maneuvers, and adenosine, and are thought to result from cAMP-mediated triggered activity.
Sinoatrial node dysfunction and deafness is a rare genetic disease characterized by congenital severe to profound deafness with no evidence of vestibular dysfunction, associated with sinoatrial node dysfunction with pronounced bradycardia and increased variability of heart rate at rest and episodic syncopes that may be triggered by enhanced physical activity and stress.
In nodular forms of adiposis dolorosa, the differential diagnosis should include other multiple lipoma syndromes such as familial multiple lipomatosis, multiple symmetric lipomatosis, myoclonic epilepsy with red ragged fibres (MERRF) syndrome, neurofibromatosis type 1, and multiple endocrine neoplasia type 1 (MEN1).
Adiposis dolorosa is a condition characterized by painful folds of fatty (adipose) tissue or the growth of multiple noncancerous (benign) fatty tumors called lipomas. This condition occurs most often in women who are overweight or obese, and signs and symptoms typically appear between ages 35 and 50. In people with adiposis dolorosa, abnormal fatty tissue or lipomas can occur anywhere on the body but are most often found on the torso, buttocks, and upper parts of the arms and legs. Lipomas usually feel like firm bumps (nodules) under the skin. The growths cause burning or aching that can be severe, particularly if they are pressing on a nearby nerve. In some people, the pain comes and goes, while in others it is continuous.
Description Adiposis dolorosa, also known as Dercum disease, is characterized by generalized obesity and pronounced, disabling, and chronic pain in the adipose tissue of the proximal extremities, trunk, pelvic area, and buttocks; the face and hands are usually spared. There are a number of associated symptoms, including multiple lipomas, generalized weakness, fatigue, sleep disturbances, constipation, and psychiatric abnormalities. It is 5 to 30 times more common in women than men, and usually presents between 35 and 50 years of age (summary by Campen et al., 2001; review by Hansson et al., 2012). Based on a review of the literature and studies of 111 patients, Hansson et al. (2012) proposed a classification of Dercum disease into 4 types: (I) generalized diffuse form without clear lipomas, (II) generalized nodular form with multiple lipomas, (III) localized nodular form, and (IV) juxtaarticular form with solitary fatty deposits near joints. Clinical Features This disorder, which was first described by Dercum (1892), is characterized by painful subcutaneous lipomas in a background of obesity.
Adiposis dolorosa Other names Anders disease Adiposis dolorosa of the diffuse truncal form (Dercum), The anterior view shows the peculiar apron of fat and the small size of the hands. The posterior view shows the arrangement of fat in folds over the hips. Specialty Endocrinology Adiposis dolorosa , is an outdated term for many years used synonymously as Dercum's disease , lipedema or Anders disease. [1] While there are numerous references to Adiposis dolorosa, it is recommended that the term no longer be used. Dercum's is now recognized as a separate condition, as is lipedema. [2] [3] References [ edit ] ^ Herbst KL. Subcutaneous Adipose Tissue Diseases: Dercum Disease, Lipedema, Familial Multiple Lipomatosis and Madelung Disease.
Adiposis dolorosa is a rare condition characterized by the growth of multiple, painful, lipomas (benign, fatty tumors). The lipomas may occur anywhere on the body and can cause severe pain. Other symptoms may include weakness, fatigue, and memory disturbances. It usually occurs in adults, and women are more commonly affected than men. Adiposis dolorosa is chronic and tends to be progressive. The exact cause is unknown.
Rather, the symptoms are alleviated through the ritual of gangshinje , a type of gut in which the mu receives her god or spirit. [4] In the fourth version of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV), published by the American Psychiatric Association , [5] shinbyeong , or shin-byung , is listed as an example of a culture-bound syndrome . Religious aspects [ edit ] In the tradition of Muism, the shinbyeong is considered a structured religious experience demonstrating the vertical connection between god and humanity and showing that "god in some form exists in human consciousness." ... The shinbyeong is dissociated from reality and enters a higher form of consciousness . [6] See also [ edit ] Mu (shaman) Muism References [ edit ] ^ Kim 1998 , pp. 41–42 ^ Kim 1998 , pp. 42–43 ^ Kim 1998 , pp. 43–44 ^ Kim 1998 , pp. 48–49 ^ American Psychiatric Association (2000). "Glossary of Culture-Bound Syndromes: shin-byung". Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) .
Mosquito allergy may result in a collection of symptoms called skeeter syndrome that occur after a bite. This syndrome may be mistaken for an infection such as cellulitis .
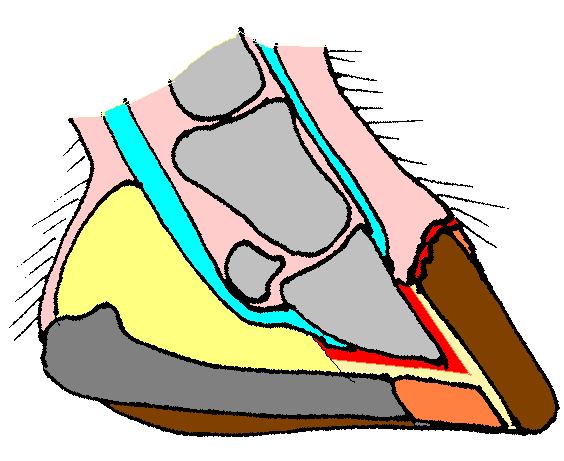
The hoof of a foal with HWSD HWSD causes the layers within the dorsal hoof wall (brown, far right) to separate from each other Hoof wall separation disease , (HWSD), is an autosomal recessive genetic hoof disease in horses . [1] Research is being carried out at, among others, UC Davis School of Veterinary Medicine in Davis in California . [2] The disease has been found in Connemara ponies and was earlier referred to as Hoof Wall Separation Syndrome , HWSS. Contents 1 Symptoms 2 Origin and cause 3 Tests and treatment 4 References 5 External links Symptoms [ edit ] The disease develops among foals from the age of one to six months and typically occurs during their first year of life. [3] The frontal edge of the hoof cracks and splits. ... Protecting the hooves from such variations may reduce the effect of the disease. [4] References [ edit ] ^ a b UC Davis Veterinary medicine: Connemara Pony Hoof Wall Separation Disease , accessed April 25, 2015 ^ a b Hoof Wall Separation Syndrome in Connemara Ponies , Accessed November 4, 2014 ^ a b c Finno, CJ; Stevens, C; Young, A; Affolter, V; Joshi, NA; Ramsay, S; Bannasch, DL (April 2015).
Fruit allergies make up about ten percent of all food related allergies. [1] Contents 1 Symptoms 2 Other symptoms due to hypersensitivity 3 Different allergic fruits 4 Diagnostic 5 Allergy or intolerance 6 Mitigation 7 See also 8 References 8.1 External references 9 External links Symptoms [ edit ] Allergic reactions to fruit and vegetables are usually mild and often just affect the mouth, causing itching, a rash, or blisters where the food touches the lips and mouth. This is called oral allergy syndrome . A number of people who react in this way to fruit or vegetables will also react to pollen from some trees and weeds. ... People suffering from allergies may suffer from a hypersensitivity to the allergic food, which is what causes the allergic reaction. Most fruit allergies are oral syndrome allergies because they are consumed but may also be an external allergy if the fruit touches the skin.
Mature T-cell lymphoma , also called Peripheral T-Cell lymphoma , is a group of rare, aggressive lymphomas that develop from mature white blood cells and originate from lymphoid tissues outside of the bone marrow. Mature T-cell lymphoma is under the category of non-Hodgkin lymphoma . Mature T-cell lymphomas account for 10% to 15% of all lymphomas and is more common in Asia than in Europe and America. [1] Its common subtypes include angioimmunoblastic T-cell lymphoma , anaplastic large cell lymphoma and peripheral T-cell lymphoma not otherwise specified . [1] While different subtypes have variable symptoms, common symptoms include enlarged painless lymph nodes , fever, weight loss, rash and night sweats. [2] Some subtypes of mature T-cell lymphoma may be associated with viral exposure [3] [4] as well as gene mutations . [5] Diagnosis is done by physical examinations, assisted by tests like biopsy , PET scan and CT scan to examine the site of lymph node development. [6] Chemotherapy , drugs, autologous stem cell treatment and extracorporeal photopheresis are treatment options. [7] The choice of treatment and its subsequent effectiveness are determined by the subtype present in the patient. Contents 1 Subtypes of Mature T-Cell Lymphoma 1.1 Angioimmunoblastic T-Cell Lymphoma 1.2 Anaplastic Large Cell Lymphoma 1.3 Peripheral T-Cell Lymphoma Not Otherwise Specified 1.4 Less Common Subtypes 2 Signs and Symptoms 3 Causes 3.1 Viral Exposure 3.2 Gene Mutations 4 Diagnosis 4.1 Biopsy 5 Treatment 5.1 Chemotherapy 5.2 Drugs 5.3 Autologous Stem Cell Treatment 5.4 Extracorporeal Photopheresis (ECPP) 6 References Subtypes of Mature T-Cell Lymphoma [ edit ] There are many different subtypes under mature T-cell lymphoma, each being considered as a separate disease due to specific clinical features. Incidence of each subtype is subjected to geographical variations. The World Health Organisation (WHO) had identified the naming and features of the subtypes in the “WHO Classification Tumours of Haematopoietic and Lymphoid Tissues”, published in 2008. [1] A revision was made in 2016 to update information obtained from advanced researches. [8] The common subtypes are angioimmunoblastic T-cell lymphoma, anaplastic large cell lymphoma and peripheral T-cell lymphoma not otherwise specified. [1] Angioimmunoblastic T-Cell Lymphoma [ edit ] Angioimmunoblastic T-cell lymphoma (AITL) is a fast-growing form of mature T-cell lymphoma, accounting for 18.5% of patients. [9] It is characterised by systemic disorders, polymorphous lymphoid infiltrate and a significant increase in proliferation of follicular dendritic cells and high endothelial venules . [10] It originates from follicular T helper (T FH ) cells , [11] which is important in maintaining immune response.
An extremely rare, primary cutaneous T-cell lymphoma disorder characterized by solitary, or multifocal and diffuse, cutaneous lesions, ranging from tumor-like patches, plaques, papules, nodules, and/or erythroderma, located on any area of the body, which rapidly progress and may become ulcerated and/or infected. Systemic involvement may be associated.
Shrinkage of the breasts Breast atrophy 15th century sculpture depicting breast atrophy Breast atrophy is the normal or spontaneous atrophy or shrinkage of the breasts . [1] Breast atrophy commonly occurs in women during menopause when estrogen levels decrease. [2] [3] [4] It can also be caused by hypoestrogenism and/or hyperandrogenism in women in general, [1] such as in antiestrogen treatment for breast cancer , in polycystic ovary syndrome (PCOS), [5] [6] and in malnutrition such as that associated with eating disorders like anorexia nervosa or with chronic disease . [1] [7] [8] It can also be an effect of weight loss . [8] [9] In the treatment of gynecomastia in males and macromastia in women, and in hormone replacement therapy (HRT) for trans men , [10] breast atrophy may be a desired effect. ... ISBN 978-0-521-88159-3 . ^ Ricardo Azziz (3 July 2007). The Polycystic Ovary Syndrome: Current Concepts on Pathogenesis and Clinical Care .