Overview Dry socket is a painful dental condition that sometimes happens after you have a tooth removed. Having a tooth removed is called an extraction. Dry socket happens when a blood clot at the site where the tooth was removed does not form, comes out or dissolves before the wound has healed. Usually a blood clot forms at the site where a tooth was removed. This blood clot is a protective layer over the underlying bone and nerve endings in the empty tooth socket. Also, the clot contains cells that are needed for proper healing of the site. Intense pain happens when the underlying bone and nerves are exposed.
Contents 1 Signs and symptoms 2 Causes 2.1 Examples 3 Mechanisms 4 Diagnosis 5 Treatments 5.1 Gene therapy prior to conception 6 Epidemiology 7 History 8 Notable cases 9 References 10 External links Signs and symptoms [ edit ] Symptoms include: poor growth loss of muscle coordination muscle weakness visual problems hearing problems learning disabilities heart disease liver disease kidney disease gastrointestinal disorders respiratory disorders neurological problems autonomic dysfunction dementia [1] Acquired conditions in which mitochondrial dysfunction has been involved are: diabetes Huntington's disease cancer Alzheimer's disease Parkinson's disease bipolar disorder , [2] [3] [4] schizophrenia , aging and senescence, anxiety disorders [5] cardiovascular disease sarcopenia chronic fatigue syndrome [3] The body, and each mutation, is modulated by other genome variants; the mutation that in one individual may cause liver disease might in another person cause a brain disorder. ... Examples [ edit ] Examples of mitochondrial diseases include: Mitochondrial myopathy Diabetes mellitus and deafness (DAD) this combination at an early age can be due to mitochondrial disease Diabetes mellitus and deafness can be found together for other reasons Leber's hereditary optic neuropathy (LHON) visual loss beginning in young adulthood eye disorder characterized by progressive loss of central vision due to degeneration of the optic nerves and retina affects 1 in 50,000 people in Finland Leigh syndrome , subacute sclerosing encephalopathy after normal development the disease usually begins late in the first year of life, although onset may occur in adulthood a rapid decline in function occurs and is marked by seizures, altered states of consciousness, dementia, ventilatory failure Neuropathy, ataxia, retinitis pigmentosa, and ptosis (NARP) progressive symptoms as described in the acronym dementia Myoneurogenic gastrointestinal encephalopathy (MNGIE) gastrointestinal pseudo-obstruction neuropathy Myoclonic Epilepsy with Ragged Red Fibers (MERRF) progressive myoclonic epilepsy "Ragged Red Fibers" are clumps of diseased mitochondria that accumulate in the sub sarcolemmal region of the muscle fiber and appear when muscle is stained with modified Gömöri trichrome stain short stature hearing loss lactic acidosis exercise intolerance MELAS syndrome Mitochondrial DNA depletion syndrome Conditions such as Friedreich's ataxia can affect the mitochondria but are not associated with mitochondrial proteins. ... Using genetic engineering in attempts to produce babies free of mitochondrial disease is controversial in some circles and raises important ethical issues . [26] [27] A male baby was born in Mexico in 2016 from a mother with Leigh syndrome using spindle transfer. [28] In September 2012 a public consultation was launched in the UK to explore the ethical issues involved. [29] Human genetic engineering was used on a small scale to allow infertile women with genetic defects in their mitochondria to have children. [30] In June 2013, the United Kingdom government agreed to develop legislation that would legalize the 'three-person IVF ' procedure as a treatment to fix or eliminate mitochondrial diseases that are passed on from mother to child. ... Charlie Gard , a British boy who suffered from mitochondrial DNA depletion syndrome ; decisions about his care were taken to various law courts.
Congenital motor nystagmus is an outdated term for IIN. Infantile nystagmus syndrome (INS) is an umbrella term used to describe different forms of infantile nystagmus (with or without sensory defects) characterized by an increasing slow phase velocity. ... Disorders to Consider in the Differential Diagnosis of FRMD7 -Related Infantile Nystagmus (FIN) View in own window DiffDx Disorder Gene(s) MOI Clinical Features of DiffDx Disorder Overlapping w/FIN Distinguishing from FIN 1 Oculocutaneous albinism (OCA) 2 OCA1 (OMIM 203100, 606952) OCA2 (OMIM 203200) OCA3 (OMIM 203290) OCA4 TYR OCA2 TYRP1 SLC45A2 AR Infantile nystagmus In OCA: Hypopigmentation of iris pigment epithelium Hypopigmentation of ocular fundus Foveal hypoplasia Misrouting of axons in optic chiasm Much poorer visual acuity (in all forms of albinism) Poor binocular vision; strabismus is common X-linked ocular albinism GPR143 XL Infantile nystagmus Normal hair & skin pigmentation X-linked inheritance Chediak-Higashi syndrome (CHS) LYST AR Infantile nystagmus In CHS: Partial OCA Immunodeficiency Mild bleeding tendency Neurologic findings during early adulthood ~85% develop the accelerated phase (lymphoproliferative infiltration of bone marrow & reticuloendothelial system) Achromatopsia AFT6 CNGA3 CNGB3 GNAT2 PDE6C PDE6H AR Infantile nystagmus (pendular or jerk nystagmus in achromatopsia) In achromatopsia: Reduced or complete loss of color discrimination 3 Reduced visual acuity Photophobia Small central scotoma Eccentric fixation Absent/markedly diminished ERG photopic response but normal/mildly abnormal scotopic response Characteristic lesion at the fovea w/outer nuclear layer thinning on optical coherence tomography Blue cone monochromatism (OMIM 303700) OPN1LW OPN1MW XL Infantile nystagmus In blue cone monochromatism: Reduced visual acuity (but better than in achromatopsia) Photophobia Reduced photopic ERG but well-preserved S cone ERG X-linked congenital stationary night blindness (CSNB1 and CSNB2) CACNA1F NYX XL Infantile nystagmus Normal color vision Normal fundus examination In CSNB: Non-progressive retinal findings of reduced visual acuity, defective dark adaptation, refractive error, & strabismus Scotopic ERG shows severely reduced (or absent) b-waves in CSNB1 & reduced but measurable b-waves in CSNB2 (absent b-wave may be referred to as a "negative ERG." ... Nystagmus in childhood can also be associated with other disorders such as aniridia, retinopathy of prematurity, dystrophies of retinal photoreceptors (including Joubert syndrome and Bardet-Biedl syndrome), congenital cataract, optic disc atrophy, and optic nerve hypoplasia. Other syndromes that can present with nystagmus during infancy include Down syndrome and spasmus nutans [Gottlob 2000].
Overview Gingivitis is a common and mild form of gum disease (periodontal disease) that causes irritation, redness and swelling (inflammation) of your gingiva, the part of your gum around the base of your teeth. It's important to take gingivitis seriously and treat it promptly. Gingivitis can lead to much more serious gum disease called periodontitis and tooth loss. The most common cause of gingivitis is poor oral hygiene. Good oral health habits, such as brushing at least twice a day, flossing daily and getting regular dental checkups, can help prevent and reverse gingivitis. Symptoms Healthy gums are firm and pale pink and fitted tightly around the teeth. Signs and symptoms of gingivitis include: Swollen or puffy gums Dusky red or dark red gums Gums that bleed easily when you brush or floss Bad breath Receding gums Tender gums When to see a dentist If you notice any signs and symptoms of gingivitis, schedule an appointment with your dentist.
Genetic Heterogeneity of Hereditary Tyrosinemia Tyrosinemia type II (TYRSN2; 276600), also known as Richner-Hanhart syndrome, is caused by mutation in the TAT gene (613018) on chromosome 16q22. ... Clinical Features Among the children of first-cousin parents, Lelong et al. (1963) observed 2 sons with cirrhosis, Fanconi renotubular syndrome, and marked increase in plasma tyrosine. ... Gentz et al. (1965) described 7 patients in 4 families with multiple renal tubular defects like those of the de Toni-Debre-Fanconi syndrome, nodular cirrhosis of the liver, and impaired tyrosine metabolism. ... In stage III, renal tubular damage (Baber syndrome), often with hypophosphatemic rickets, appears. ... Mice homozygous for certain chromosome 7 deletions that include Fah die perinatally as a result of liver dysfunction and exhibit a complex syndrome characterized by structural abnormalities and alterations in gene expression in the liver and kidney.
Tyrosinemia type 1 (HTI) is an inborn error of tyrosine catabolism caused by defective activity of fumarylacetoacetate hydrolase (FAH) and is characterized by progressive liver disease, renal tubular dysfunction, porphyria-like crises and a dramatic improvement in prognosis following treatment with nitisinone. Epidemiology Birth incidence is 1/100,000 in most areas but is more common is some regions, notably in Québec, Canada. Clinical description HT1 is clinically heterogenous. Symptoms may start during the first few months (acute type), in second half of the first year (subacute type) or in the following years up to adulthood (chronic type). In the acute type, manifestations of hepatic failure predominate (bleeding diathesis, hypoglycemia, ascites etc) with frequent sepsis and rapid deterioration. Mild proximal tubular disease is usually present. Subacute type manifests a similar but less severe clinical picture presenting usually with hepatomegaly or hypophosphatemic rickets (due to tubular dysfunction).
Tyrosinemia type 1 is a genetic disorder characterized by elevated blood levels of the amino acid tyrosine, a building block of most proteins. This condition is caused by a shortage of the enzyme fumarylacetoacetate hydrolase, one of the enzymes required for the multi-step process that breaks down tyrosine. This enzyme shortage is caused by mutations in the FAH gene. Symptoms usually appear in the first few months of life and include failure to thrive , diarrhea, vomiting, jaundice, cabbage-like odor, and increased tendency to bleed (particularly nosebleeds). Tyrosinemia type I can lead to liver and kidney failure, softening and weakening of the bones, problems affecting the nervous system, and an increased risk of liver cancer. This condition is inherited in an autosomal recessive manner. Treatment should be started as soon as the condition is diagnosed and includes a diet restricted in tyrosine and phenylalanine along with nitisinone, a medication that blocks the second step in the tyrosine degradation pathway.
Prevention of secondary complications: Treatment of early signs of carnitine deficiency, osteoporosis, and rickets that are secondary to renal tubular Fanconi syndrome. Surveillance: Guidelines for routine surveillance of individuals with tyrosinemia type I have been established. ... The renal tubular dysfunction involves a Fanconi-like renal syndrome with generalized aminoaciduria, phosphate loss, and, for many, renal tubular acidosis. ... Differential Diagnosis of Tyrosinemia Type I in Infants by Presenting Finding View in own window Presenting Finding Differential Diagnosis Hypertyrosinemia Immature liver High-protein diet 1, 2 Tyrosinemia type II (OMIM 276600) Tyrosinemia type III (OMIM 276710) Other liver disease Hypermethioninemia Homocystinuria Disorders of methionine metabolism Other liver disease Liver disease Galactosemia Hereditary fructose intolerance Fructose 1, 6 biphosphatase deficiency (OMIM 229700) Niemann-Pick disease type C Wilson disease Neonatal hemochromatosis (OMIM 231100) Hemophagocytic lymphohistiocytosis Mitochondrial cytopathies Congenital disorders of glycosylation Transaldolase deficiency (OMIM 606003) Acetaminophen toxicity Bacterial infections (sepsis, salmonella, TB) Viral infections (e.g., CMV, hepatitis A/B, herpes) Mushroom poisoning 3 Herbal medicines 3 Idiosyncratic drug reaction, toxin, vascular/ischemic or infiltrative process 3 Renal syndrome Lowe syndrome Cystinosis Renal tubular acidosis Fanconi syndrome Rickets Hypophosphatasia Vitamin D deficiency (nutritional/genetic) Hypophosphatemic rickets Vitamin D-dependent rickets Fanconi syndrome Neurologic crisis Cerebral hemorrhage/edema Bacterial/viral meningitis Hypernatremic dehydration Acute intermittent porphyria 1. ... Prevention of Secondary Complications Because carnitine deficiency secondary to the renal tubular Fanconi syndrome can cause skeletal muscle weakness, serum concentration of carnitine should be measured so that carnitine deficiency, if identified, can be treated [Nissenkorn et al 2001].
Tyrosinemia is a genetic disorder characterized by disruptions in the multistep process that breaks down the amino acid tyrosine, a building block of most proteins. If untreated, tyrosine and its byproducts build up in tissues and organs, which can lead to serious health problems. There are three types of tyrosinemia, which are each distinguished by their symptoms and genetic cause. Tyrosinemia type I, the most severe form of this disorder, is characterized by signs and symptoms that begin in the first few months of life. Affected infants fail to gain weight and grow at the expected rate (failure to thrive) due to poor food tolerance because high-protein foods lead to diarrhea and vomiting.
Synofzik et al [2014] concluded that the three PRPS1- related phenotypes (CMTX5, Arts syndrome, and DFNX1) constitute a continuum after observing all three phenotypes in one family with a loss-of-function pathogenic variant: a male with CMT and Arts syndrome and a heterozygous female with hearing loss due to skewing of X-chromosome inactivation. ... Genotype-Phenotype Correlations The established PRPS1 -related disorders are not distinct entities, but rather clusters on a phenotypic continuum as evidenced by overlap of the features of CMTX5 / Arts syndrome / DFNX1 both in affected individuals and within families.
Namburi et al. (2016) noted that the phenotype observed in these patients did not fall within the standard classification of Usher syndrome (see 276900), in which the retinal disease is typical retinitis pigmentosa with early rod dysfunction. ... Fu et al. (2017) designated the phenotype as an atypical form of Usher syndrome. Mapping In a consanguineous Jewish family with cone-rod dystrophy and sensorineural hearing loss, Namburi et al. (2016) performed homozygosity mapping and identified a 65.6-Mb region on chromosome 9, as well as a 34-Mb region on chromosome 19. ... The authors also screened 71 unsolved cases of Usher syndrome, but did not find any biallelic CEP78 mutations.
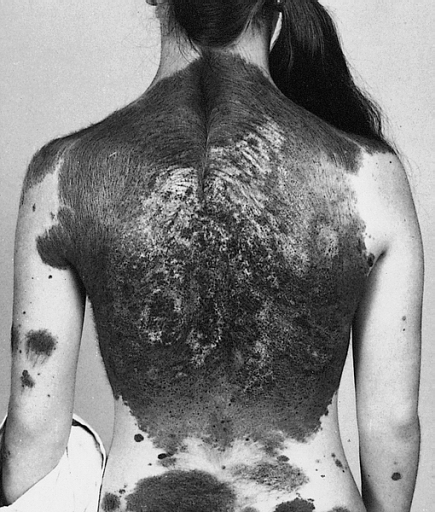
Congenital disorder involving melanocytic tumours in the skin and brain Neurocutaneous melanosis Other names Neurocutaneous melanosis syndrome [1] A 13-year-old with a giant congenital melanocytic nevus, or "bathing trunk" Neurocutaneous melanosis is a congenital disorder characterized by the presence of congenital melanocytic nevi on the skin and melanocytic tumors in the leptomeninges of the central nervous system . [2] These lesions may occur in the amygdala , cerebellum , cerebrum , pons and spinal cord of patients. ... These symptoms seem to be present regardless of the malignancy of the melanin deposits within the central nervous system. [7] Approximately 10% of patient with neurocutaneous melanosis also present the Dandy–Walker syndrome and associated Dandy-Walker malformation. ... Therefore, few symptomatic cases (around 100) have been reported to date. [10] See also [ edit ] Dandy–Walker syndrome Melanoma Phakomatosis References [ edit ] ^ "Neurocutaneous melanosis | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov .
The disorder is a rare but severe manifestation of congenital melanocytic nevus syndrome (CMNS; 137550). Some patients with neurocutaneous melanosis or CMNS may develop malignant melanoma. ... Molecular Genetics Kinsler et al. (2013) identified somatic oncogenic missense mutations affecting codon 61 of the NRAS gene in affected cutaneous and neurologic tissues from 12 of 15 patients with congenital melanocytic nevus syndrome and/or neurocutaneous melanosis.
Neurocutaneous melanocytosis (NCM) is a rare congenital neurological disorder characterized by abnormal aggregations of nevomelanocytes within the central nervous system (leptomeningeal melanocytosis) associated with large or giant congenital melanocytic nevi (CMN; see this term). NCM can be asymptomatic or present as variably severe and progressive neurological impairment, sometimes resulting in death. Epidemiology Prevalence is estimated at 1/50,000-1/200,000. The incidence of symptomatic NCM appears to be approximately a third to a half of these. Clinical description A large, or giant, CMN is a pigmented skin lesion of more than 20 cm projected adult diameter (40 for "giant"), composed of aggregated melanocytes in a delimited area of the body, and presenting with an elevated risk of malignant transformation. Leptomeningeal melanocytosis nearly always presents with CMN, though not conversely; a case with no pigmented lesions and another with only café-au-lait spots have been reported.
Neurocutaneous melanosis (NCM) is a rare, non-inherited condition of the central nervous system. It is characterized by melanocytic nevi in both the skin and the brain. Two-thirds of people with NCM have giant congenital melanocytic nevi, and the remaining one-third have numerous lesions but no giant lesions. The typical cutaneous lesions are present at birth. Neurological features typically present in the first or second year. Intracranial hypertension is the most common presentation, along with seizures, decreased alertness, and cranial nerve dysfunction.The underlying cause, while not completely understood, is believed to be a primary defect in the neural crest.
Mutation in the B3GALT6 gene also causes a spondylodysplastic type of Ehlers-Danlos syndrome (EDSSPD2; 615349), which has overlapping features with SEMDJL1. ... Some also had a range of extraskeletal and connective tissue abnormalities that overlapped with those seen in the progeroid form of Ehlers-Danlos syndrome (see EDSP2, 615349). Malfait et al. (2013) described affected members of 3 unrelated Iranian families with a severe autosomal recessive disorder characterized by skin fragility, delayed wound healing, joint hyperlaxity and contractures, muscle hypotonia, intellectual disability, and a spondyloepimetaphyseal dysplasia with bone fragility, multiple early-onset fractures, and severe kyphoscoliosis. The authors described the disorder as a 'pleiotropic Ehlers-Danlos-syndrome-like connective tissue disorder.'
A number sign (#) is used with this entry because of evidence that spondyloepimetaphyseal dysplasia with joint laxity-3 (SEMDJL3) is caused by homozygous mutation in the EXOC6B gene (607880) on chromosome 2p13. Description Spondyloepimetaphyseal dysplasia with joint laxity-3 (SEMDJL3) is characterized by multiple joint dislocations at birth, severe joint laxity, scoliosis, gracile metacarpals and metatarsals, delayed bone age, and poorly ossified carpal and tarsal bones (Girisha et al., 2016). For a discussion of genetic heterogeneity of SEMD with joint laxity, see SEMDJL1 (271640). Clinical Features Spranger et al. (2006) described 5 patients with a 'leptodactylic' form of SEMD, including 2 sisters (patients 2 and 3) who were later found to have a mutation in the EXOC6B gene (see MOLECULAR GENETICS). Both sisters were born with bilateral hip and patellar dislocations. Examination at ages 12 and 9.5 years, respectively, showed similar findings.
A rare primary bone dysplasia characterized by short stature, joint laxity, vertebral anomalies, severe progressive spinal malalignment leading to spinal cord compression, progressive kyphoscoliosis, thoracic asymmetry, and elbow and foot deformities. Additional features include mild skin hyperelasticity, spatulate terminal phalanges, cleft palate and lip, structural cardiac malformations, and mild facial dysmorphism (oval face, prominent eyes with blue sclerae, and a long upper lip).
B-cell lymphoma refers to types of non-Hodgkin lymphoma that are characterized by abnormalities of the "B-cells" (a type of white blood cell that makes antibodies to help fight infection). The condition may grow and spread slowly with few symptoms (also known as indolent lymphoma) or may be very aggressive with severe symptoms. When present, signs and symptoms may include swollen lymph nodes in the neck, armpit, or groin; abdominal pain; fatigue; fever; night sweats; and/or weight loss. The underlying cause of B-cell lymphoma is poorly understood. However, the condition can be associated with genetic abnormalities, environmental factors, viruses, immunodeficiency states, and connective-tissue disorders . Treatment is based on many factors, including the severity of the condition and the associated signs and symptoms.
A disorder that caused by the Junin virus (JUNV), is an acute viral hemorrhagic disease characterized by initial fever and malaise followed by gastrointestinal symptoms and in some cases hemorrhagic and neurological manifestations.
The patient’s older brother, father, and paternal uncle had previously all died of ALS or an ALS type syndrome. The patient developed progressive bulbar palsy, became dependent on a respirator, and had two episodes of cardiac arrest . ... Oxford University Press. (2000) External links [ edit ] Classification D ICD - 10 : G12.2 ICD - 10-CM : G12.22 ICD - 9-CM : 335.22 MeSH : D010244 06-095b. at Merck Manual of Diagnosis and Therapy Home Edition v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis
Progressive bulbar palsy involves the brain stem. The brain stem is the part of the brain needed for swallowing, speaking, chewing, and other functions. Signs and symptoms of progressive bulbar palsy include difficulty swallowing, weak jaw and facial muscles, progressive loss of speech, and weakening of the tongue. Additional symptoms include less prominent weakness in the arms and legs, and outbursts of laughing or crying (called emotional lability). Progressive bulbar palsy is considered a variant form of amyotrophic lateral sclerosis (ALS). Many people with progressive bulbar palsy later develop ALS. While there is no cure for progressive bulbar palsy or for ALS, doctors can treat symptoms.