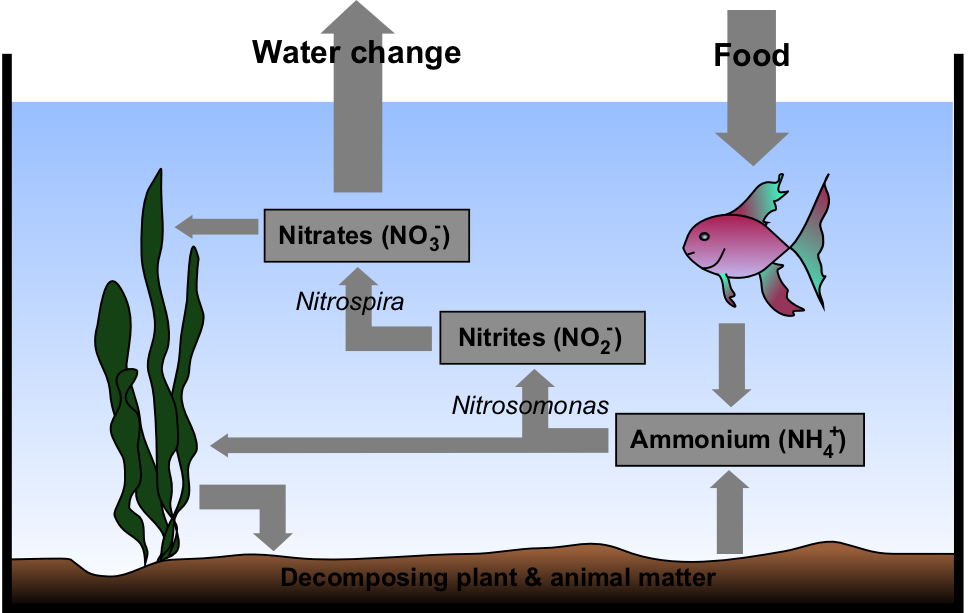
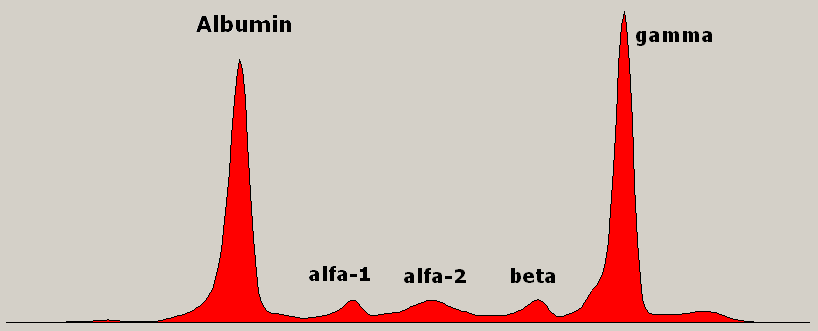
Rappaport et al. (1968) observed 2 brothers with a salt-losing syndrome due to decreased aldosterone. The authors postulated a defect in 18-OH-dehydrogenase. ... Rosler et al. (1973, 1977) reported a syndrome of profound salt wastage in 12 children from 8 Iranian Jewish families.
Lymph node biopsy was positive for EBV, consistent with a diagnosis of EBV-induced B-cell lymphoproliferative syndrome. She was treated with chemotherapy, but died of complications 4 months after bone marrow transplantation. ... INHERITANCE - Autosomal recessive HEAD & NECK Mouth - Oral thrush (in 1 patient) RESPIRATORY - Recurrent respiratory infections NEUROLOGIC Central Nervous System - Delayed psychomotor development (in some patients) Behavioral Psychiatric Manifestations - Attention-deficit disorder (in some patients) - Hyperactivity (in some patients) IMMUNOLOGY - Primary immunodeficiency - Lymphopenia - Decreased numbers of naive CD4+ and CD8+ T cells - Decreased circulating CD4+ T cells - Impaired survival of mature T cells - Decreased invariant NK T cells - Post-vaccination varicella (in 1 patient) - Low to normal immunoglobulin levels - Variable antibody response to vaccination - Mildly decreased or normal T-cell proliferative responses - Normal thymus - Increased susceptibility to EBV infection NEOPLASIA - EBV-induced B-cell lymphoma LABORATORY ABNORMALITIES - Shortened telomeres (1 patient) MISCELLANEOUS - Onset in infancy or childhood - Mucocutaneous immunodeficiency syndrome may be prominent - Three unrelated families have been reported (last curated October 2014) MOLECULAR BASIS - Caused by mutation in the coronin 1A gene (CORO1A, 605000.0001 ) ▲ Close
Brenner tumor of the ovary is a solid, abnormal growth (tumor) on the ovary. Most Brenner tumors are not cancerous (benign). About 5% of Brenner tumors are cancerous (malignant) or have a small chance of spreading beyond its original location (borderline). These tumors most often occur in women after menopause. They usually do not cause symptoms unless they are very large. When symptoms are present, they may include abdominal pain or vaginal bleeding. The cause of Brenner tumors is unknown. They are usually found by accident during surgery for another reason or during a routine doctor's examination.
This condition is less common in other countries, but the incidence is unknown. [5] Even though this disease can occur in various races and ethnicities, another study backed this finding up by stating that 1 in 26,000 people in Finland had the disease and that 1 in 18,000 were carriers. [3] After trisomy 21 and fragile X syndrome , this is the most frequent multiple congenital anomaly/intellectual disability syndrome in Finland. [7] See also [ edit ] Inborn error of metabolism References [ edit ] ^ "Aspartylglycosaminuria | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program" . rarediseases.info.nih.gov .
External links [ edit ] Classification D ICD - 10 : L90.4 ICD - 9-CM : 701.8 DiseasesDB : 32940 External resources eMedicine : derm/4 v t e Cutaneous keratosis, ulcer, atrophy, and necrobiosis Epidermal thickening keratoderma : Keratoderma climactericum Paraneoplastic keratoderma Acrokeratosis paraneoplastica of Bazex Aquagenic keratoderma Drug-induced keratoderma psoriasis Keratoderma blennorrhagicum keratosis : Seborrheic keratosis Clonal seborrheic keratosis Common seborrheic keratosis Irritated seborrheic keratosis Seborrheic keratosis with squamous atypia Reticulated seborrheic keratosis Dermatosis papulosa nigra Keratosis punctata of the palmar creases other hyperkeratosis : Acanthosis nigricans Confluent and reticulated papillomatosis Callus Ichthyosis acquisita Arsenical keratosis Chronic scar keratosis Hyperkeratosis lenticularis perstans Hydrocarbon keratosis Hyperkeratosis of the nipple and areola Inverted follicular keratosis Lichenoid keratosis Multiple minute digitate hyperkeratosis PUVA keratosis Reactional keratosis Stucco keratosis Thermal keratosis Viral keratosis Warty dyskeratoma Waxy keratosis of childhood other hypertrophy: Keloid Hypertrophic scar Cutis verticis gyrata Necrobiosis / granuloma Necrobiotic/palisading Granuloma annulare Perforating Generalized Subcutaneous Granuloma annulare in HIV disease Localized granuloma annulare Patch-type granuloma annulare Necrobiosis lipoidica Annular elastolytic giant-cell granuloma Granuloma multiforme Necrobiotic xanthogranuloma Palisaded neutrophilic and granulomatous dermatitis Rheumatoid nodulosis Interstitial granulomatous dermatitis / Interstitial granulomatous drug reaction Foreign body granuloma Beryllium granuloma Mercury granuloma Silica granuloma Silicone granuloma Zirconium granuloma Soot tattoo Tattoo Carbon stain Other/ungrouped eosinophilic dermatosis Granuloma faciale Dermis / localized CTD Cutaneous lupus erythematosus chronic: Discoid Panniculitis subacute : Neonatal ungrouped: Chilblain Lupus erythematosus–lichen planus overlap syndrome Tumid Verrucous Rowell's syndrome Scleroderma / Morphea Localized scleroderma Localized morphea Morphea–lichen sclerosus et atrophicus overlap Generalized morphea Atrophoderma of Pasini and Pierini Pansclerotic morphea Morphea profunda Linear scleroderma Atrophic / atrophoderma Lichen sclerosus Anetoderma Schweninger–Buzzi anetoderma Jadassohn–Pellizzari anetoderma Atrophoderma of Pasini and Pierini Acrodermatitis chronica atrophicans Semicircular lipoatrophy Follicular atrophoderma Linear atrophoderma of Moulin Perforating Kyrle disease Reactive perforating collagenosis Elastosis perforans serpiginosa Perforating folliculitis Acquired perforating dermatosis Skin ulcer Pyoderma gangrenosum Other Calcinosis cutis Sclerodactyly Poikiloderma vasculare atrophicans Ainhum / Pseudo-ainhum v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
There is a possible association between ultrasound-detected fetal CPCs and Trisomy 18 . [5] [6] It is not correlated to the presence of Trisomy 21 ( Down syndrome ). [7] [8] Generally the risks are very low if there are no other risk factors. ... "Choroid plexus cysts: not associated with Down syndrome". Ultrasound in Obstetrics and Gynecology . 8 (4): 232–235. doi : 10.1046/j.1469-0705.1996.08040232.x .
A developmental disorder affecting premature infants, likely secondary to an immaturity of respiratory control resulting in idiopathic pauses in breathing often associated with reduced heart rate and arterial blood oxygen levels. It may be exacerbated by concurrent neonatal diseases.
(The latter report concerned association with Rieger syndrome (180500).) Poskitt and Rayner (1974) described 2 families, each with a father and son affected by isolated GH deficiency. ... In addition to short stature, the craniofacial features of 18p monosomy may resemble those of Turner syndrome: round face, hypertelorism, flattened nasal bridge, and wide mouth with small upper lip.
The disorder is distinct from juvenile pernicious anemia due to selective intestinal malabsorption of vitamin B12 with proteinuria (261100) and pernicious anemia associated with the polyglandular autoimmune syndrome (240300). It is also distinct from classic adult-onset pernicious anemia (170900). ... In 7 families previously diagnosed with Imerslund-Grasbeck syndrome (261100) due to inconclusive results on radiocobalamin absorption tests, but who were negative for mutations in the cubilin (CUBN; 602997) or the AMN (605799) gene, Tanner et al. (2005) identified homozygosity for 6 different mutations in the GIF gene (609342.0002-609342.0007).
Differential diagnosis Differential diagnosis includes Imerslund-Gräsbeck syndrome, transcobalamin II deficiency, cblF defect, and acquired pernicious anemia (see these terms), which is caused by autoimmunity or Helicobacter infection.
In the 14.5-year-old son of a first-cousin Algerian couple, Zittoun et al. (1988) identified deficiency of both intrinsic factor and R binder. Separate deficiencies are well described (see 261000 and 193090, respectively). The boy presented with severe megaloblastic anemia, growth retardation, and neurologic dysfunction with typical features of subacute combined degeneration of the spinal cord. The anemia responded completely to cyanocobalamin and folic acid. Intrinsic factor was absent from gastric juice, but acid secretion and gastric mucosa were normal. R binders were absent from gastric juice as well as from serum, saliva, and polymorphonuclear leukocytes.
Intrinsic factor deficiency is a rare condition that is characterized by pernicious anemia and neurological abnormalities. Most affected people develop signs and symptoms of the condition before age 5 years which may include failure to thrive and symptoms related to anemia (i.e. fatigue, pale skin, etc). Without early diagnosis and treatment, nervous system damage may occur which can be associated with confusion; depression; and numbness or tingling in the hands and/or feet. Intrinsic factor deficiency is caused by changes (mutations) in the GIF gene and is inherited in an autosomal recessive manner. Treatment generally consists of vitamin B12 injections.
Malignant tumors of the vascular system are relatively uncommon and usually have an unfavorable, rapid clinical course. Metastases usually occur early by hematogenous routes to the lung and bone as well as to the regional lymph nodes. These vascular tumors may develop anywhere and can be difficult to diagnose, both clinically and histologically. Histogenetically they are of 2 main types: malignant hemangioendotheliomas originating from intimal endothelial cells, and malignant hemangiopericytomas arising from adventitial perivascular cells or pericytes. Plukker et al. (1988) reported malignant hemangiopericytoma in a brother and sister, aged 19 and 22 years, respectively, and in a 25-year-old fifth cousin of theirs.
Hemangiopericytoma is a term used to described a group of tumors that are derived from pericytes, the cells normally arranged along specific types of blood vessels called capillaries and venules. These types of tumors are typically slow-growing, may be either benign (non-cancerous) or malignant (cancerous), and may occur anywhere in the body.
The different forms of IIP that can be associated with FPF are as follows (listed in order of relative frequency): Idiopathic pulmonary fibrosis (IPF)/cryptogenic fibrosing alveolitis (CFA) Nonspecific interstitial pneumonitis (NSIP) Cryptogenic organizing pneumonia (COP) Acute interstitial pneumonia (AIP; also called Hamman-Rich syndrome) Respiratory bronchiolitis associated interstitial lung disease (RB-ILD) Desquamative interstitial pneumonitis (DIP) Lymphocytic interstitial pneumonitis (LIP) Idiopathic pulmonary fibrosis (IPF) is the most common form of IIP and is present in approximately 55% of families with FPF. ... Approaches to molecular genetic testing can include: Serial single-gene testing of TERT , TERC , SFTPC , and SFTPA2 based on family history, age of onset, phenotypes suggestive of a telomerase syndrome, or the presence of lung cancer: TERT and TERC account for the majority of families with a pathogenic variant and may or may not show clinical features of a "telomerase syndrome" as described below. ... Other inherited disorders that exhibit diffuse parenchymal lung disease as a clinical feature: Hermansky-Pudlak syndrome (HPS) is a multisystem disorder characterized by tyrosinase-positive oculocutaneous albinism, a bleeding diathesis resulting from a platelet storage pool deficiency, and, in some cases, pulmonary fibrosis or granulomatous colitis. Pulmonary fibrosis typically causes symptoms in the early thirties and progresses to death within a decade. Hermansky-Pudlak syndrome is known to be associated with a defect in five-hydroxytryptamine and lysosomal metabolism. In Hermansky-Pudlak syndrome, the release of PDGF-B by alveolar macrophages is enhanced.
Nomenclature In the past, the following terms were used to describe XLP1: Epstein-Barr virus infection, familial fatal EBV susceptibility (EBVS) X-linked progressive combined variable immunodeficiency 5 Purtilo syndrome Duncan disease Prevalence The estimated prevalence of XLP is one per one million males. ... To date, lymphoma as a complication of XLP2 has not been reported. Chediak-Higashi syndrome is characterized by partial oculocutaneous albinism, a mild bleeding tendency, and severe immunodeficiency. ... Inheritance is autosomal recessive. Griscelli syndrome type 2 (GS2; OMIM 607624) is a disorder of cytotoxic T lymphocytes caused by mutation of RAB27A , encoding a small GTPase, which controls the movement of vesicles within cells [Ménasché et al 2002]. ... ITK deficiency (lymphoproliferative syndrome 1; OMIM 613011). Pathogenic variants in ITK have been reported in association with an autosomal recessive form of lymphoproliferative disease [Huck et al 2009, Stepensky et al 2011]. ... CD27 deficiency (lymphoproliferative syndrome 2; OMIM 615122). Pathogenic variants in TNFRSF7 have been reported in association with an autosomal recessive form of lymphoproliferative disease [van Montfrans et al 2012, Salzer et al 2013].
X-linked lymphoproliferative disease (XLP) is a disorder of the immune system and blood-forming cells that is found almost exclusively in males. More than half of individuals with this disorder experience an exaggerated immune response to the Epstein-Barr virus (EBV). EBV is a very common virus that eventually infects most humans. In some people it causes infectious mononucleosis (commonly known as "mono"). Normally, after initial infection, EBV remains in certain immune system cells (lymphocytes) called B cells. However, the virus is generally inactive (latent) because it is controlled by other lymphocytes called T cells that specifically target EBV-infected B cells.
A rare, genetic, primary immunodeficiency disorder characterized by an abnormal immune response to Epstein-Barr virus (EBV) infection, caused by hemizygous mutations in the X-linked SH2D1A gene, resulting in B cell lymphoproliferation and manifesting with various phenotypes which include EBV-driven severe or fulminant mononucleosis, hemophagocytic lymphohistiocytosis (presenting with fulminant hepatitis, hepatic necrosis, bone marrow hypoplasia, and neurological involvement), hypogammaglobulinemia, and B-cell lymphoma. Additional variable manifestations include vasculitis, lymphomatoid granulomatosis, aplastic anemia, and chronic gastritis. Occasionally, T-cell lymphoma may be observed. Laboratory findings include normal or increased activated T cells and reduced memory B cells.
Autosomal Dominant Disorders with Macrocephaly and Intellectual Disability to Consider in the Differential Diagnosis of EED -Related Overgrowth View in own window DiffDx Disorder Gene Clinical Features of the DiffDx Disorder Overlapping w/ EED overgrowth Distinguishing from EED overgrowth EZH2 -related overgrowth (includes Weaver syndrome) EZH2 Macrosomia Craniofacial dysmorphism Advanced bone age Cancer predisposition Less frequent cervical spine anomalies SUZ12 -related overgrowth 1 SUZ12 Postnatal overgrowth Craniofacial dysmorphism Advanced bone age Prenatal overgrowth apparently less severe Postnatal overgrowth apparently more severe Sotos syndrome NSD1 Macrosomia Craniofacial dysmorphism Advanced bone age Cancer predisposition Malan syndrome (OMIM 614753) NFIX Macrosomia Craniofacial dysmorphism Advanced bone age Cancer predisposition Less frequent cervical spine anomalies HIST1H1E syndrome HIST1H1E Neonatal hypoglycemia Camptodactyly Kyphoscoliosis Neonatal hypertonia Beckwith-Wiedemann syndrome CDKN1C Macrosomia Neonatal hypoglycemia Umbilical hernia Cancer predisposition Organomegaly Macroglossia PTEN hamartoma tumor syndrome PTEN Macrocephaly Intellectual disability Cancer predisposition Mucocutaneous lesions DiffDx = differential diagnosis 1.
Other (more rarely) associated behaviors can include: Hand flapping Agitation Motor and/or vocal tics (Tourette syndrome) Hyperventilation episodes Teeth grinding (bruxism) Attention-deficit disorder (ADD) Pica Self-harm behaviors including self-biting, face scratching, and head banging Onset of self-injurious behavior can be as early as age two years; some individuals display this behavior later in life. ... Nomenclature ASXL3 -related disorder was first described by Bainbridge et al [2013] in four unrelated individuals with truncating variants in ASXL3 ; it is sometimes referred to as Bainbridge-Ropers syndrome. Prevalence The prevalence of ASXL3 -related disorder is not known. ... See OMIM Phenotypic Series: Autosomal dominant ID; Autosomal recessive ID; Nonsyndromic X-linked ID; and Syndromic X-linked ID. Note: Heterozygous pathogenic variants ASXL1 and ASXL2 , the other two genes in the ASXL gene family (see Molecular Pathogenesis), are associated with Bohring-Opitz syndrome (BOS) and Shashi-Pena syndrome, respectively. Both disorders are characterized by developmental delay but can be distinguished from ASXL3 -related disorder by the characteristic facial dysmorphism associated with BOS, and by macrocephaly and abnormal brain imaging in Shashi-Pena syndrome. Management Consensus clinical management guidelines for ASXL3 -related disorder have not been published.
A rare, neurodegenerative, autosomal recessive human disease causing severe disability Ataxia–telangiectasia Other names Louis–Bar syndrome Specialty Neurology , medical genetics Ataxia–telangiectasia ( AT or A–T ), also referred to as ataxia–telangiectasia syndrome or Louis–Bar syndrome , [1] is a rare, neurodegenerative , autosomal recessive disease causing severe disability. ... Characteristics of the ATM protein [3] [27] [28] [29] [30] [31] [32] [33] [34] A–T has been described as a genome instability syndrome, a DNA repair disorder and a DNA damage response (DDR) syndrome. ... These include: Ataxia–oculomotor apraxia type 1 (AOA1) Ataxia–oculomotor apraxia type 2 (AOA2 also known as SCAR1) Ataxia–telangiectasia like disorder (ATLD) Nijmegen breakage syndrome (NBS) Comparison of clinical and laboratory features of rare genetic disorders than can be confused with A–T Ataxia–oculomotor apraxia type 1 (AOA1) is an autosomal recessive disorder similar to A–T in manifesting increasing problems with coordination and oculomotor apraxia, often at a similar age to those having A–T. ... Because those mutations of Mre11 that severely impair the MRE11 protein are incompatible with life, individuals with ATLD all have some partial function of the Mre11 protein, and hence likely all have their own levels of disease severity. Nijmegen breakage syndrome (NBS) is a rare genetic disorder that has similar chromosomal instability to that seen in people with A–T, but the problems experienced are quite different. ... These studies further demonstrated that the treatment is well tolerated, with a good safety profile. [ citation needed ] A multinational clinical trial investigating N-Acetyl-L-Leucine for the treatment Ataxia-Telangiectasia began in 2019. [81] IntraBio is also conducting two parallel clinical trials with N-Acetyl-L-Leucine for the treatment Niemann-Pick disease type C [82] and GM2 Gangliosidosis ( Tay-Sachs and Sandhoff Disease) [83] Future opportunities to develop N-Acetyl-Leucine include Lewy Body Dementia , [84] Amyotrophic lateral sclerosis , Restless Leg Syndrome , Multiple Sclerosis , and Migraine . [85] Prognosis [ edit ] The life expectancy of people with A–T is highly variable.
A rare, genetic, persistent combined dystonia characterized by clinical signs similar to ataxia-telangiectasia but with a later (usually adulthood) onset and slower progression. Patients typically present extrapyramidal signs, such as resting tremor, choreathetosis, and dystonia, as the initial symptoms and later often develop mild cerebellar ataxia (with gait usually preserved). Telangiectasia and immunodeficiency may be absent but secondary features of ataxia-telangiectasia, such as risk of malignancy, dysarthria and peripheral neuropathy, are frequently present.