Rare X-linked dominant condition Nance–Horan syndrome Other names Cataract X-linked with Hutchinsonian teeth This condition is inherited in an X-linked dominant manner. Specialty Ophthalmology Nance–Horan syndrome is a rare X linked syndrome characterized by congenital cataract leading to profound vision loss, characteristic dysmorphic features and dental anomalies. [1] [2] Microcornea, microphthalmia and mild or moderate mental retardation may accompany these features. ... You can help by adding to it . ( August 2017 ) Management [ edit ] There is no known cure for this syndrome. Patients usually need ophthalmic surgery and may also need dental surgery.Genetic counseling and screening of the mother's relatives is recommended. [ citation needed ] History [ edit ] This syndrome was first described by Margaret B. ... PMID 2246772 . ^ Bixler, D; Higgins, M; Hartsfield Jr, J (1984). "The Nance-Horan syndrome: A rare X-linked ocular-dental trait with expression in heterozygous females". ... "A Turkish family with Nance-Horan syndrome due to a novel mutation". Gene . 525 (1): 141–145. doi : 10.1016/j.gene.2013.03.094 .
A number sign (#) is used with this entry because Nance-Horan syndrome (NHS) is caused by mutation in the NHS gene (300457) on chromosome Xp22. ... Another family with the Nance-Horan syndrome was reported by van Dorp and Delleman (1979). ... Some members of the family had mild to moderate nonocular clinical features suggestive of Nance-Horan syndrome. By fine mapping, the authors localized the disease gene to Xp22.13. ... The clinical features were consistent with both Nance-Horan syndrome and early infantile epileptic encephalopathy-2 (EIEE2; 300672), which is caused by mutation in the CDKL5 gene. Liao et al. (2011) reported 2 Taiwanese brothers with a clinical diagnosis of Nance-Horan syndrome associated with a 915-kb deletion at chromosome Xp22.13 inherited from their mother.
Nance-Horan syndrome (NHS) is characterized by the association in male patients of congenital cataracts with microcornea, dental anomalies and facial dysmorphism. ... Differential diagnosis Differential diagnosis includes: X-linked microphthalmia, Lenz syndrome, Oculo-facio-cardio-dental (OFCD) syndrome, and Oculo-cerebro-renal (Lowe) syndrome (see these terms).
Nance-Horan syndrome is a rare genetic disorder that may be evident at birth. ... The range and severity of symptoms may vary greatly from one person to another, even among affected members of the same family. Nance-Horan syndrome is caused by a mutation in the NHS gene and is inherited as an X-linked dominant trait, which means that both males and females can be affected, but males often have more severe symptoms.The treatment is directed toward the specific symptoms that are apparent in the individual.
S2CID 36002793 . ^ NCBI PubMed; Bentley TP; Brennan DF (August 2009). "Lemierre's syndrome: methicillin-resistant Staphylococcus aureus (MRSA) finds a new home". ... PMID 18280087 . ^ a b c d Puymirat E, Biais M, Camou F, Lefèvre J, Guisset O, Gabinski C (March 2008). "A Lemierre's syndrome variant caused by Staphylococcus aureus ". ... PMID 24152679 . ^ Screaton NJ, Ravenel JG, Lehner PJ, Heitzman ER, Flower CD (November 1999). "Lemierre Syndrome: Forgotten but Not Extinct-Report of Four Cases" . ... "Human necrobacillosis, with emphasis on Lemierre's syndrome" . Clinical Infectious Diseases . 31 (2): 524–532. doi : 10.1086/313970 . ... PMID 32445216 . ^ UK Chief Medical Officer Update 29 February 2001 ( CMO Update29 Feb 2001 ) ^ Valerio, Luca; Corsi, Gabriele (2020). "Lemierre syndrome: Current evidence and rationale of the Bacteria-Associated Thrombosis, Thrombophlebitis and LEmierre syndrome (BATTLE) registry" .
Ichthyosis prematurity syndrome Specialty Dermatology Ichthyosis prematurity syndrome ( IPS ) is a dermatological disease with known genetic causes. ... Premature birth [ edit ] Pregnancies that have a foetus affected with this syndrome are complicated because of polyhydramnion . ... Eosionphelia is an abnormal increase of eosinophils in tissue, blood or both and is present in individuals born with this syndrome. [7] Genetics [ edit ] Mode of inheritance [ edit ] This rare syndrome is associated with an autosomal recessive mode of inheritance. ... "Assignment of the locus for ichthyosis prematurity syndrome to chromosome 9q33.3-34.13" . J Med Genet . 41 (3): 208–212. doi : 10.1136/jmg.2003.012567 . ... PMID 14985385 . ^ a b c "Ichthyosis Prematurity Syndrome" . OMIM . Retrieved 3 December 2015 . ^ Ikuya, Tsuge; Masashi, Morishita; Takema, Kato; et al. (2015).
Ichthyosis prematurity syndrome is a rare, syndromic congenital ichthyosis characterized by premature birth (at gestational weeks 30-32, in general) in addition to thick, caseous and desquamating epidermis, neonatal respiratory asphyxia, and persistent eosinophilia.
A number sign (#) is used with this entry because ichthyosis prematurity syndrome (IPS) is caused by mutation in the FATP4 (SLC27A4; 604194) gene. ... The rare subtype ichthyosis prematurity syndrome presents with complications at mid-trimester of pregnancy leading to prematurity, a thick caseous and desquamating skin, respiratory complications, and persistent eosinophilia. ... Mapping Klar et al. (2004) performed genomewide linkage analysis in 16 families with ichthyosis prematurity syndrome from Norway and Sweden. Thirteen families had 1 or 2 affected members or had pairs of sibs with at least 1 affected; 3 families had 1 affected single child. ... In 28 affected and 22 healthy sibs from 22 unrelated Norwegian and Swedish families with ichthyosis prematurity syndrome, 13 of which had been previously studied by Klar et al. (2004), Melin et al. (2006) confirmed linkage to chromosome 9q33.3-q34.13 and refined the IPS haplotype to a 76-kb core region. ... Pathogenesis Klar et al. (2009) observed abnormal distribution of lipids between epidermal layers in ichthyosis prematurity syndrome that is consistent with the expression pattern of FATP4 in normal epidermis, and suggested a defect in lipid homeostasis and skin barrier formation in IPS.
A contiguous gene syndrome comprising otodental syndrome (characterized by globodontia and sensorineural high-frequency hearing deficit) associated with eye abnormalities including, typically, iris and chorioretinal coloboma, as well as, on occasion, microcornea, microphtalmos, lenticular opacity, lens coloboma and iris pigment epithelial atrophy.
Ocular coloboma segregating with otodental syndrome has been reported (summary by Gregory-Evans et al., 2007). Clinical Features Levin et al. (1975) and Levin and Jorgenson (1972, 1974) described a syndrome of sensorineural hearing loss and dental anomalies in a 6-generation kindred of Italian ancestry. ... The authors noted that Witkop et al. (1976) had observed that patients with otodental syndrome had long facies, anteverted nostrils, long philtrum and a full-cheek appearance. ... Molecular Genetics In a Brazilian ('OD1') and a Belgian ('OD3') family with otodental syndrome, originally described by de Toledo et al. (1971) and Van Doorne et al. (1998), respectively, and in a British family with otodental syndrome and coloboma ('OD2'), originally reported by Winter (1983), Gregory-Evans et al. (2007) identified overlapping hemizygous microdeletions on chromosome 11q13 by SNP and Southern blot analysis. ... Gregory-Evans et al. (2007) suggested that FGF3 haploinsufficiency is likely the cause of otodental syndrome and that FADD haploinsufficiency accounts for the associated ocular coloboma.
Ulnar/fibula ray defect - brachydactyly syndrome is a very rare malformation syndrome characterized by ulnar hypoplasia associated with hypoplastic to absent fourth and/or fifth digits, fibular hypoplasia, short stature and facial dysmorphism.
Two-point linkage analysis with microsatellite markers spanning the ulnar-mammary syndrome (181450) locus at 12q24.1 did not confirm linkage. Morava et al. (2003) suggested that the patients in this family may have a previously undescribed syndrome. INHERITANCE - Autosomal dominant GROWTH Height - Short stature HEAD & NECK Face - Midface hypoplasia - Round face - Prominent forehead CARDIOVASCULAR Heart - Atrial septal defect CHEST Breasts - Normal breast SKELETAL Limbs - Unilateral ulnar hypoplasia - Fibular hypoplasia - Leg length discrepancy Hands - Brachydactyly - Postaxial oligodactyly (absent 4-5 fingers) Feet - Bilateral clubfeet - Syndactyly (4-5 toes) SKIN, NAILS, & HAIR Skin - Normal sweating - Hemangioma (neck and forehead) ▲ Close
Intellectual disability-spasticity-ectrodactyly syndrome is a rare intellectual disability syndrome characterized by severe intellectual disability, spastic paraplegia (with wasting of the lower limbs) and distal transverse defects of the limbs (e.g. ectrodactyly, syndactyly, clinodactyly of the hands and/or feet).
Jancar (1967) reported the case of a 19-year-old man with ectrodactyly, mental retardation, and spastic paraplegia. Zlotogora (1987) described the combination in a 3-year-old child. Zlotogora and Glick (1993) reported a third case in an offspring of healthy Moslem Arabs who were first cousins. They raised the possibility of autosomal recessive inheritance because the parents of 2 of the patients were first cousins and the sister of patient 2 may also have been affected. However, the second and third families originated from communities in which consanguineous marriages are the rule and not the exception. Limbs - Ectrodactyly Neuro - Mental retardation - Spastic paraplegia Inheritance - Autosomal recessive ▲ Close
Microlissencephaly-micromelia syndrome is a syndrome of abnormal cortical development, characterized by severe prenatal polyhydramnios, postnatal microcephaly, lissencephaly, upper limb micromelia, dysmorphic facies (coarse face, hypertrichosis, and short nose with long philtrum), intractable seizures, and early death.
Polydactyly-myopia syndrome is an exceedingly rare autosomal dominant developmental anomaly reported in 1986 in nine individuals among four generations of the same family. The syndrome is characterized clinically by four-limb postaxial polydactyly and progressive myopia.
Polydactyly myopia syndrome is characterized by postaxial polydactyly (the presence of an extra digit on the side of the hand or foot by the pinky or small toe) and progressive myopia .
Czeizel and Brooser (1986) found a disorder of postaxial polydactyly and progressive myopia in 9 persons in 4 generations of a family in an autosomal dominant pedigree pattern. There was no instance of male-to-male transmission. The proband also had bilateral congenital inguinal hernias and undescended testes but the former condition came from the other side of the family. Limbs - Postaxial polydactyly Eyes - Progressive myopia Inheritance - Autosomal dominant ▲ Close
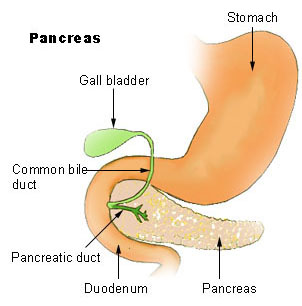
Johanson-Blizzard syndrome Other names JBS The pancreas , and its location within the digestive system . ... "Case report. Johanson-Blizzard syndrome: a report of gender-discordant twins with a novel UBR1 mutation" . ... S2CID 20598132 . ^ a b c d e Rosanowski F, Hoppe U, Hies T, Eysholdt U (Oct 1998). "Johanson-Blizzard syndrome. A complex dysplasia syndrome with aplasia of the nasal alae and inner ear deafness". ... "Pathophysiology of the pancreatic defect in Johanson-Blizzard syndrome: a disorder of acinar development". ... "Johanson-Blizzardov sindrom" [The Johanson-Blizzard syndrome]. Lijec Vjesn (in Croatian). 120 (5): 114–6.
A number sign (#) is used with this entry because of evidence that Johanson-Blizzard syndrome (JBS) is caused by homozygous or compound heterozygous mutation in the UBR1 gene (605981) on chromosome 15q15. ... Nagashima et al. (1993) described diabetes mellitus, first detected at age 11 years, in a girl with Johanson-Blizzard syndrome. Vanlieferinghen et al. (2001) described a case of Johanson-Blizzard syndrome in a neonate. ... Vanlieferinghen et al. (2003) described the prenatal ultrasonographic diagnosis of a recurrence of Johanson-Blizzard syndrome in a subsequent pregnancy in this family. ... Elting et al. (2008) reported 2 unrelated girls with a mild form of Johanson-Blizzard syndrome, born of Turkish and Iranian consanguineous parents, respectively. ... Inheritance The possibility of X-linked dominance lethal in the male was raised by Konigsmark and Gorlin (1976) since most patients had been female and the syndrome may have been observed in an XXY male.
Whilst atherosclerosis of spinal arteries is rare, necrosis (death of tissue) in the anterior artery can be caused by disease in vessels originating from the segmental arteries such as atheroma (arterial wall swelling) or aortic dissection (a tear in the aorta ). [3] Contents 1 Spinal cord infarction 1.1 Anterior spinal artery syndrome 1.2 Posterior spinal artery syndrome 1.3 Transient ischemic attack 2 Spinal arteriovenous malformations 3 See also 4 References 5 External links Spinal cord infarction [ edit ] Anterior spinal artery syndrome [ edit ] Main article: Anterior spinal artery syndrome Anterior spinal artery syndrome is necrosis of tissue in the anterior spinal artery or its branches. [4] It is characterised by pain which radiates at onset and sudden quadraplegia (paralysis of all four limbs) or paraplegia (paralysis of the lower body). ... The prognosis is dependent upon individual circumstances and factors. [3] Posterior spinal artery syndrome [ edit ] Posterior spinal artery syndrome is much rarer than its anterior counterpart as the white matter structures that are present are much less vulnerable to ischemia since they have a better blood supply. When posterior spinal artery syndrome does occur, dorsal columns are damaged and ischemia may spread into the posterior horns. ... ISBN 0-443-04345-0 . ^ Manconi M, Mondini S, Fabiani A, Rossi P, Ambrosetto P, Cirignotta F (December 2003). "Anterior spinal artery syndrome complicated by the ondine curse" . ... External links [ edit ] Classification D ICD - 10 : G95.1 ICD - 9-CM : 336.1 MeSH : D020758 v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis v t e Focal lesions of the spinal cord General Myelopathy Myelitis Spinal cord compression By location Brown-Séquard syndrome Posterior cord syndrome Anterior cord syndrome Central cord syndrome Cauda equina syndrome Other Polio Demyelinating disease Transverse myelitis Tropical spastic paraparesis Epidural abscess Syringomyelia Syringobulbia Morvan's syndrome Sensory ataxia Tabes dorsalis Abadie's sign Subacute combined degeneration of spinal cord Vascular myelopathy Anterior spinal artery syndrome Foix–Alajouanine syndrome
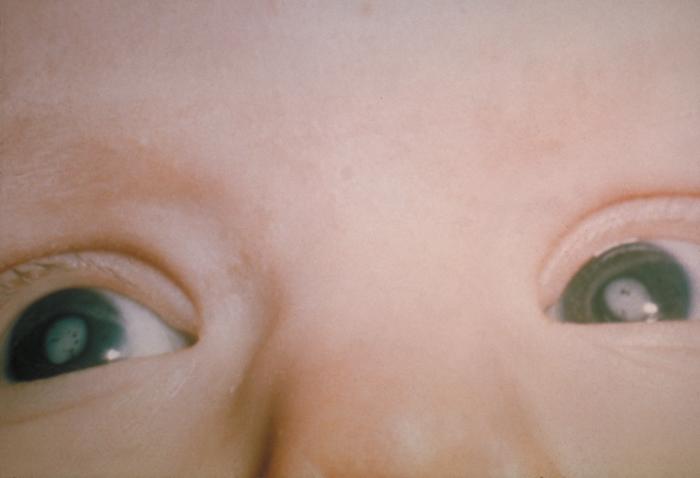
Find sources: "Congenital rubella syndrome" – news · newspapers · books · scholar · JSTOR ( December 2007 ) ( Learn how and when to remove this template message ) Congenital rubella syndrome White pupils due to congenital cataracts in a child with congenital rubella syndrome Specialty Teratology Congenital rubella syndrome ( CRS ) can occur in a developing fetus of a pregnant woman who has contracted rubella , usually in the first trimester. ... It was discovered in 1941 by Australian Norman McAlister Gregg . [1] Contents 1 Signs and symptoms 2 Prevention 3 References 4 External links Signs and symptoms [ edit ] Infant with skin lesions from congenital rubella "Salt-and-pepper" retinopathy is characteristic of congenital rubella. [2] Congenital rubella serology time-line The classic triad for congenital rubella syndrome is: [3] Sensorineural deafness (58% of patients) Eye abnormalities—especially retinopathy , cataract , glaucoma , and microphthalmia (43% of patients) Congenital heart disease —especially pulmonary artery stenosis and patent ductus arteriosus (50% of patients) [4] Other manifestations of CRS may include: Spleen , liver , or bone marrow problems (some of which may disappear shortly after birth) Intellectual disability Small head size ( microcephaly ) Low birth weight [5] Thrombocytopenic purpura Extramedullary hematopoiesis (presents as a characteristic blueberry muffin rash ) Enlarged liver Small jaw size Skin lesions [5] Children who have been exposed to rubella in the womb should also be watched closely as they age for any indication of: Developmental delay [5] Autism [6] Schizophrenia [7] Growth retardation [8] Learning disabilities Diabetes mellitus [9] Prevention [ edit ] Vaccinating the majority of the population is effective at preventing congenital rubella syndrome. [10] For women who plan to become pregnant, the MMR (measles mumps, rubella) vaccination is highly recommended, at least 28 days prior to conception. [5] The vaccine should not be given to women who are already pregnant as it contains live viral particles. [5] Other preventative actions can include the screening and vaccinations of high-risk personnel, such as medical and child care professions. [11] References [ edit ] ^ Atkinson, William (2011). ... PMID 20029144 . ^ "Congenital rubella syndrome | Sense" . www.sense.org.uk . Retrieved 2015-07-30 . ^ Oster ME, Riehle-Colarusso T, Correa A (January 2010). "An update on cardiovascular malformations in congenital rubella syndrome" . Birth Defects Research Part A: Clinical and Molecular Teratology . 88 (1): 1–8. doi : 10.1002/bdra.20621 . ... External links [ edit ] Classification D ICD - 10 : P35.0 ICD - 9-CM : 771.0 MeSH : D012410 DiseasesDB : 11729 External resources MedlinePlus : 001658 eMedicine : emerg/388 v t e Skin infections , symptoms and signs related to viruses DNA virus Herpesviridae Alpha HSV Herpes simplex Herpetic whitlow Herpes gladiatorum Herpes simplex keratitis Herpetic sycosis Neonatal herpes simplex Herpes genitalis Herpes labialis Eczema herpeticum Herpetiform esophagitis Herpes B virus B virus infection VZV Chickenpox Herpes zoster Herpes zoster oticus Ophthalmic zoster Disseminated herpes zoster Zoster-associated pain Modified varicella-like syndrome Beta Human herpesvirus 6 / Roseolovirus Exanthema subitum Roseola vaccinia Cytomegalic inclusion disease Gamma KSHV Kaposi's sarcoma Poxviridae Ortho Variola Smallpox Alastrim MoxV Monkeypox CPXV Cowpox VV Vaccinia Generalized vaccinia Eczema vaccinatum Progressive vaccinia Buffalopox Para Farmyard pox : Milker's nodule Bovine papular stomatitis Pseudocowpox Orf Sealpox Other Yatapoxvirus : Tanapox Yaba monkey tumor virus MCV Molluscum contagiosum Papillomaviridae HPV Wart / plantar wart Heck's disease Genital wart giant Laryngeal papillomatosis Butcher's wart Bowenoid papulosis Epidermodysplasia verruciformis Verruca plana Pigmented wart Verrucae palmares et plantares BPV Equine sarcoid Parvoviridae Parvovirus B19 Erythema infectiosum Reticulocytopenia Papular purpuric gloves and socks syndrome Polyomaviridae Merkel cell polyomavirus Merkel cell carcinoma RNA virus Paramyxoviridae MeV Measles Togaviridae Rubella virus Rubella Congenital rubella syndrome ("German measles" ) Alphavirus infection Chikungunya fever Picornaviridae CAV Hand, foot, and mouth disease Herpangina FMDV Foot-and-mouth disease Boston exanthem disease Ungrouped Asymmetric periflexural exanthem of childhood Post-vaccination follicular eruption Lipschütz ulcer Eruptive pseudoangiomatosis Viral-associated trichodysplasia Gianotti–Crosti syndrome v t e Conditions originating in the perinatal period / fetal disease Maternal factors complicating pregnancy, labour or delivery placenta Placenta praevia Placental insufficiency Twin-to-twin transfusion syndrome chorion / amnion Chorioamnionitis umbilical cord Umbilical cord prolapse Nuchal cord Single umbilical artery presentation Breech birth Asynclitism Shoulder presentation Growth Small for gestational age / Large for gestational age Preterm birth / Postterm pregnancy Intrauterine growth restriction Birth trauma scalp Cephalohematoma Chignon Caput succedaneum Subgaleal hemorrhage Brachial plexus injury Erb's palsy Klumpke paralysis Affected systems Respiratory Intrauterine hypoxia Infant respiratory distress syndrome Transient tachypnea of the newborn Meconium aspiration syndrome Pleural disease Pneumothorax Pneumomediastinum Wilson–Mikity syndrome Bronchopulmonary dysplasia Cardiovascular Pneumopericardium Persistent fetal circulation Bleeding and hematologic disease Vitamin K deficiency bleeding HDN ABO Anti-Kell Rh c Rh D Rh E Hydrops fetalis Hyperbilirubinemia Kernicterus Neonatal jaundice Velamentous cord insertion Intraventricular hemorrhage Germinal matrix hemorrhage Anemia of prematurity Gastrointestinal Ileus Necrotizing enterocolitis Meconium peritonitis Integument and thermoregulation Erythema toxicum Sclerema neonatorum Nervous system Perinatal asphyxia Periventricular leukomalacia Musculoskeletal Gray baby syndrome muscle tone Congenital hypertonia Congenital hypotonia Infections Vertically transmitted infection Neonatal infection rubella herpes simplex mycoplasma hominis ureaplasma urealyticum Omphalitis Neonatal sepsis Group B streptococcal infection Neonatal conjunctivitis Other Miscarriage Perinatal mortality Stillbirth Infant mortality Neonatal withdrawal v t e Vertically transmitted infections Gestational Viruses Congenital rubella syndrome Congenital cytomegalovirus infection Neonatal herpes simplex Hepatitis B Congenital varicella syndrome HIV Fifth disease Bacteria Congenital syphilis Other Toxoplasmosis transplacental TORCH complex During birth transcervical Candidiasis Gonorrhea Listeriosis Late pregnancy Listeriosis Congenital cytomegalovirus infection By breastfeeding Breastfeeding Tuberculosis HIV
Congenital rubella refers to the group of birth defects that occur in an infant whose mother is infected with the virus that causes German measles (rubella) during pregnancy. Congenital rubella occurs when the rubella virus in the mother affects the developing baby in the first 3 months of pregnancy. After the fourth month, if the mother has a rubella infection, it is less likely to harm the developing baby. The most common problems are hearing loss due to damage to the nerve pathways from the inner ear to the brain (sensorineural hearing loss), ocular abnormalities (cataract, infantile glaucoma, and pigmentary retinopathy ) and heart problems. Other symptoms and signs may include intrauterine growth retardation, prematurity , stillbirth, miscarriage, neurological problems (intellectual disability, low muscle tone, very small head), liver and spleen enlargement (hepatosplenomegaly), jaundice, skin problems, anemia, hormonal problems, and other issues.
Treatments and lifestyle adjustments can help you reduce or manage the signs and symptoms of premenstrual syndrome. Symptoms The list of potential signs and symptoms for premenstrual syndrome is long, but most women only experience a few of these problems. ... But a small number of women with premenstrual syndrome have disabling symptoms every month. ... Signs and symptoms of premenstrual syndrome change with hormonal fluctuations and disappear with pregnancy and menopause. ... Some women with severe premenstrual syndrome have undiagnosed depression, though depression alone does not cause all of the symptoms Diagnosis There are no unique physical findings or lab tests to positively diagnose premenstrual syndrome. Your doctor may attribute a particular symptom to premenstrual syndrome (PMS) if it's part of your predictable premenstrual pattern.
The presence of normal brain imaging and physical growth distinguished this syndrome from other syndromes with overlapping abnormalities. ... Chodirker et al. (2002) considered the patients reported by Khalifa et al. (2002) to have the same disorder as that in the 8 aboriginal Canadians from Manitoba and Ontario reported as having Ritscher-Schinzel syndrome (RSS; 220210) by Marles et al. (1995). Thus they believed that the debate was not about whether or not the cases described by Khalifa et al. (2002) represented a 'new' syndrome, but whether or not the cases described in these 2 reports should be considered to have RSS or a separate 'new' syndrome first reported by Marles et al. (1995). In a rebuttal, Khalifa and Cappon (2002) argued that the patients of Marles et al. (1995) were phenotypically distinct from those they reported in the same ethnic group; furthermore, they questioned the diagnosis of RSS in their own patients because they did not fulfill the criteria for RSS, also known as the 3C syndrome because of cerebellar, cardiac, and characteristic craniofacial anomalies.
Complement component 2 deficiency is a disorder that causes the immune system to malfunction, resulting in a form of immunodeficiency. Immunodeficiencies are conditions in which the immune system is not able to protect the body effectively from foreign invaders such as bacteria and viruses. People with complement component 2 deficiency have a significantly increased risk of recurrent bacterial infections, specifically of the lungs (pneumonia), the membrane covering the brain and spinal cord (meningitis), and the blood (sepsis ), which may be life-threatening. These infections most commonly occur in infancy and childhood and become less frequent in adolescence and adulthood. Complement component 2 deficiency is also associated with an increased risk of developing autoimmune disorders such as systemic lupus erythematosus (SLE) or vasculitis.
Complement component 2 deficiency (C2D) is a genetic condition that affects the immune system. Signs and symptoms include recurrent bacterial infections and risk for a variety of autoimmune conditions . Infections can be very serious and are common in early life. They become less frequent during the teen and adult years. The most frequent autoimmune conditions associated with C2D are lupus (10-20%) and vasculitis. C2D is caused by mutations in the C2 gene and is inherited in an autosomal recessive fashion.
A number sign (#) is used with this entry because of evidence that spontaneous ovarian hyperstimulation syndrome (OHSS) is caused by heterozygous mutation in the follicle-stimulating hormone receptor gene (FSHR; 136435) on chromosome 2p16. Clinical Features Ovarian hyperstimulation syndrome most often occurs as an iatrogenic complication of ovarian stimulation treatments for in vitro fertilization; the incidence of severe forms ranges from 0.5 to 5% (Delvigne and Rozenberg, 2002). ... Pathogenesis Kaiser (2003) reviewed the pathogenesis of ovarian hyperstimulation syndrome and the mechanism by which mutations in the FSH receptor result in the disorder, with explanatory diagrams. Molecular Genetics In a 25-year-old woman of Moroccan origin who had 4 of 5 spontaneous pregnancies complicated by ovarian hyperstimulation syndrome, Vasseur et al. (2003) identified heterozygosity for a mutation in the FSHR gene (T449I; 136435.0008). ... In a woman with spontaneous ovarian hyperstimulation syndrome, previously described by Olatunbosun et al. (1996), Smits et al. (2003) identified a heterozygous missense mutation in the FSHR gene (D567N; 136435.0009).
Overview Ovarian hyperstimulation syndrome is an exaggerated response to excess hormones. ... Diagnosis Ovarian hyperstimulation syndrome diagnosis may be based on: A physical exam. ... Treatment Ovarian hyperstimulation syndrome generally resolves on its own within a week or two or somewhat longer if you're pregnant. ... Self care If you develop mild ovarian hyperstimulation syndrome, you'll probably be able to continue your day-to-day routine. ... What kind of tests do I need? Does ovarian hyperstimulation syndrome usually go away on its own, or will I need treatment?
Severity ranges from mild to life-threatening and is complicated by increased risk of thrombosis, acute hepato-renal failure, acute respiratory distress syndrome, and ovarian torsion and rupture.
Ovarian hyperstimulation syndrome Specialty Gynecology Ovarian hyperstimulation syndrome ( OHSS ) is a medical condition that can occur in some women who take fertility medication to stimulate egg growth, and in other women in very rare cases. ... Retrieved 2015-07-09 . ^ Ovarian hyperstimulation syndrome Updated by: Linda J. Vorvick and Susan Storck Update. ... PMID 27577848 . ^ Ovarian Hyperstimulation Syndrome~treatment at eMedicine Further reading [ edit ] Delvigne A, Rozenberg S (2002). "Epidemiology and prevention of ovarian hyperstimulation syndrome (OHSS): a review" . Hum Reprod Update . 8 (6): 559–77. doi : 10.1093/humupd/8.6.559 . ... "Review of clinical course and treatment of ovarian hyperstimulation syndrome (OHSS)" . Hum Reprod Update . 9 (1): 77–96. doi : 10.1093/humupd/dmg005 .
Cleft palate-lateral synechia syndrome (CPLS) is a congenital malformation syndrome characterized by the association of cleft palate and intra-oral lateral synechiae connecting the free borders of the palate and the floor of the mouth. ... Differential diagnosis Phenotypic features of CLPS can occur with other congenital anomalies, particularly the Van der Woude (VDW), orofaciodigital syndrome and popliteal pterygium syndrome (see these terms). In one reported family with CPLS, the monozygotic twin of the index case had classic phenotypic features of Fryns syndrome (see this term) suggesting that CPLS may represent a mild phenotypic expression of this syndrome.
Clinical Features Fuhrmann et al. (1972) described a new syndrome of cleft palate combined with multiple cordlike adhesions between the free borders of the palate and lateral parts of the tongue and floor of the mouth. The full syndrome occurred in 5 persons, a sixth had cleft palate only, and an unaffected male transmitted the disorder to 2 children with different mothers. The disorder was distinct from the ankyloglosson superius syndrome (106280). Arshad and Goh (1994) reported 2 unrelated cases of hypoglossia congenita with anterior maxillomandibular fusion. ... They suspected that this is the same disorder as that reported by Fuhrmann et al. (1972). Gorlin (1982) saw the syndrome in a father and son. Affected families reported by Fuhrmann et al. (1972), Gassner et al. (1979), and Gorlin (1982) suggested autosomal dominant inheritance, whereas the patient reported by Villanueva-Garcia et al. (2009) was born of consanguineous parents, suggesting autosomal recessive inheritance.
Syngnathia is a congenital adhesion of the maxilla and mandible by fibrous bands. [1] References [ edit ] ^ Parkins, G. E.; Boamah, M. O. (2009). "Congenital maxillomandibular syngnathia: Case report". Journal of Cranio-Maxillofacial Surgery . 37 (5): 276–278. doi : 10.1016/j.jcms.2009.01.001 . PMID 19231229 . This article about a congenital malformation is a stub . You can help Wikipedia by expanding it . v t e
Unsourced material may be challenged and removed. Find sources: "Menke-Hennekam syndrome" – news · newspapers · books · scholar · JSTOR ( March 2019 ) ( Learn how and when to remove this template message ) This article includes a list of general references , but it remains largely unverified because it lacks sufficient corresponding inline citations . ... Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( June 2019 ) ( Learn how and when to remove this template message ) Menke-Hennekam syndrome Autosomal dominant pattern is the inheritance manner of this condition Specialty Medical genetics Causes Mutations in the CREBBP gene Menke-Hennekam syndrome is a rare condition characterised by a constellation of lesions mostly involving the brain. ... Pathopysiology [ edit ] This is not understood. Diagnosis [ edit ] This syndrome may be suspected on clinical grounds. ... Differential diagnosis [ edit ] Rubinstein-Taybi syndrome Treatment [ edit ] There is no specific treatment for this condition. ... History [ edit ] This condition was first described in 2019. [1] References [ edit ] ^ Banka S, Sayer R, Breen C, Barton S, Pavaine J, Sheppard SE, Bedoukian E, Skraban C, Cuddapah VA, Clayton-Smith J (2019) Genotype-phenotype specificity in Menke-Hennekam syndrome caused by missense variants in exon 30 or 31 of CREBBP.
Chen et al. (1982) reported 2 isolated cases of a lethal, Larsen-like multiple joint dislocation syndrome. Death occurred shortly after birth with pulmonary insufficiency due to tracheomalacia and/or lung hypoplasia. ... Mostello et al. (1991) provided the first evidence of recessive inheritance of the lethal variant of Larsen syndrome. It is not certain, of course, that this disorder is produced by mutation in a gene distinct from that of the recessive form (245600) or, for that matter, that of the dominant form (150250) of Larsen syndrome. ... Abnormal palmar creases and laryngotracheomalacia, features seen in patients with Larsen syndrome who survive, were observed in these lethal cases. Yamaguchi et al. (1996) described neuropathologic evidence of a disturbance of neuroblast migration in a 3.5-year-old girl with a Larsen-like syndrome who manifested unusually severe neurologic signs that included intractable partial seizures, tetraplegia, and psychomotor retardation. ... The authors speculated that the brain dysplasia, which had been described in previous Laresen-like syndrome cases (e.g., Clayton-Smith and Donnai, 1988, Chen et al. (1982)), may have been the result of systemic hypoxic-ischemic insults during the second half of gestation, but they did not rule out genetic factors.
A rare developmental defect with connective tissue involvement characterized by multiple joint dislocations, flattened facial appearance, abnormal palmar creases, laryngotracheomalacia, and pulmonary hypoplasia. Additional signs may include a bifid tongue, micrognathia, non-immune hydrops fetalis, and brain dysplasia. The disease is lethal shortly after birth due to respiratory insufficiency.
Rare psychological or neurological condition Athymhormic syndrome Specialty Psychiatry Athymhormic syndrome (from Ancient Greek θυμός thūmós , "mood" or "affect", and hormḗ , "impulse", "drive" or "appetite"), psychic akinesia , or auto-activation deficit (AAD) is a rare psychopathological and neurological syndrome characterized by extreme passivity, apathy , blunted affect and a profound generalized loss of self-motivation and conscious thought. ... This “kinetic blockade” disappeared instantaneously when his son told him to move. [1] The existence of such symptoms in patients after damage to certain structures in the brain has been used in support of a physical model of motivation in human beings, wherein the limbic loop of the basal ganglia is the initiator of directed action and thought. [2] First described by French neurologist Dominique Laplane in 1982 as "PAP syndrome" ( French : perte d'auto-activation psychique , or "loss of psychic autoactivation"), the syndrome is believed to be due to damage to areas of the basal ganglia or frontal cortex , specifically the striatum and globus pallidus , responsible for motivation and executive functions. [3] It may occur without any preexisting psychiatric condition. ... "Auto‐Activation deficit: A basal ganglia related syndrome". Mov. Disord . 16 (5): 810–814. doi : 10.1002/mds.1185 .
A number sign (#) is used with this entry because of evidence that the IFAP syndrome with or without BRESHECK syndrome is caused by mutation in the MBTPS2 (300294) gene on chromosome Xp22. ... Clinical Features Early Reports The syndrome of ichthyosis follicularis with atrichia and photophobia (IFAP syndrome) was first described by MacLeod (1909), who reported a family in which 3 of 5 boys were affected. ... The 2 syndromes can be differentiated mainly on the basis of skeletal and intestinal anomalies present in the dermotrichic syndrome and ocular and respiratory disorders in the IFAP syndrome. ... Photophobia and recurrent respiratory infections are characteristic of the IFAP syndrome; nail anomalies, hypohidrosis, megacolon, and vertebral defects are characteristic of dermotrichic syndrome. ... Wang et al. (2014) designated the phenotype in their patient as 'IFAP with Olmsted syndrome-like features' and questioned whether X-linked Olmsted syndrome represented an independent condition or a severe form of IFAP.
X-linked mental retardation, Reish type is characterised by Brain anomalies, severe mental Retardation, Ectodermal dysplasia, Skeletal deformities (vertebral anomalies, scoliosis, polydactyly), Ear/eye anomalies (maldevelopment, small optic nerves, low set and large ears with hearing loss) and Kidney dysplasia/hypoplasia (giving the acronym BRESEK syndrome). Epidemiology It has been described in two brothers, one of whom died shortly after birth. Clinical description One of the brothers also had Hirschsprung disease and Cleft palate/cryptorchidism (giving the acronym: BRESHECK syndrome) Genetic counseling Transmission is X-linked dominant.