Rudiger syndrome Rudiger syndrome is inherited in an autosomal recessive manner Rudiger syndrome is a congenital disorder characterized by the association of severe growth retardation with abnormalities of the extremities, urogenital abnormalities and facial abnormalities. [1] It has been described in a family where an affected brother and sister died as infants. [2] Both autosomal recessive and autosomal dominant inheritance have been suggested with the disorder. [1] [3] The features ectrodactyly , ectodermal dysplasia and cleft palate have been described with Rudiger syndrome, giving it the rarely used designation "EEC syndrome". [3] However, this is not to be confused with the formal EEC syndrome associated with chromosome 7 . [4] It was characterized in 1971. [5] References [ edit ] ^ a b "Orphanet: Rudiger syndrome" . ... (Feb 1978). "Rudiger (E. E. C.) syndrome: report of a case associated with atopic dermatitis (author's transl)".
Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( December 2016 ) Odontoma dysphagia syndrome Other names Odontomatosis-aortae esophagus stenosis syndrome Odontoma dysphagia syndrome (Bader syndrome) is a rare syndrome (<10 cases reported to date) first described in 1967. [1] The cause is not known but it is suspected to be genetic in origin. Contents 1 Presentation 2 Cause 3 Treatment 4 References 5 External links Presentation [ edit ] This syndrome is characterized by clustering of teeth (compound odontoma, dysplasia and aplasia of teeth), slight craniofacial abnormalities, and dysphagia . ... Oral Surg Oral Med OralPathol 23:770 ^ Schönberger W (1974) Multiple odontomas (Odontomatosis) and dysphagia in father and son-a syndromic connection? Z Kinderheilkd 117(2):101-108 ^ Ziebart T, Draenert FG, Galetzka D, Babaryka G, Schmidseder R, Wagner W, Bartsch O (2012) The original family revisited after 37 years: odontoma-dysphagia syndrome is most likely caused by a microduplication of chromosome 11q13.3, including the FGF3 and FGF4 genes.
Schonberger (1974) described severe dysphagia in father and son with multiple odontomas. An isolated case reported by Boder (1967) had the same combination. Hypertrophy of the smooth muscles of the esophagus was thought to be the cause of the dysphagia. Gorlin (1977) stated that the same family was reported by Schmidseder and Hausamen (1973). Mouth - Odontoma Lab - Esophageal smooth muscle hypertrophy GI - Dysphagia Inheritance - Autosomal dominant ▲ Close
In some patients this syndrome has also been associated with Sjögren's syndrome and hemolytic anemia . ... This disease may cause white or yellow-ish spots on the arms or legs. The syndrome, a special case of scleroderma, is named after the American physician, Telfer B. ... References [ edit ] ^ "OMIM Entry - # 613471 - REYNOLDS SYNDROME" . omim.org . Retrieved 6 August 2017 . ... Peters: Primary biliary cirrhosis with scleroderma, Raynaud's phenomenon and telangiectasia. New syndrome. American Journal of Medicine, New York, 1971, 50 (3): 302–312. Volker Stadie, Johannes Wohlrab, Wolfgang Christian Marsch: The Reynolds Syndrome - a Rare Combination of Two Autoimmune Diseases.
Reynolds syndrome (RS) is an autoimmune disorder characterized by the association of primary biliary cirrhosis (PBC) with limited cutaneous systemic sclerosis (lcSSc) (see these terms). ... In some, RS is associated with other autoimmune diseases such as Sjögren's syndrome, autoimmune hemolytic anemia and (in one case) with thymoma (see these terms). An overlap syndrome between nodular regenerative hyperplasia of the liver (see this term), PBC, and lcSSc may exist.
Reynolds syndrome is an autoimmune disease characterized by the co-occurrence of primary biliary cholangitis (PBC) and limited cutaneous systemic sclerosis (LCSS). The signs and symptoms of Reynolds syndrome therefore include those of both PBC and LCSS. ... Signs and symptoms of LCSS may include calcium deposits in the skin, tissues, and organs (calcinosis); sores on the fingers and toes ( digital ulcers ); facial telangiectasias ; Raynaud's phenomenon ; esophageal dysfunction (such as acid reflux); and sclerodactyly (tightening of the skin on the fingers and toes). Reynolds syndrome typically occurs sporadically, affecting only one person in a family.
A number sign (#) is used with this entry because of evidence that Reynolds syndrome is caused by heterozygous mutation in the LBR gene (600024) on chromosome 1q42. ... These findings were reminiscent of CREST syndrome (see 181750). Three patients had upper gastrointestinal bleeding, which was related to telangiectasia or esophageal varices. ... Gaudy-Marqueste et al. (2010) reported a 76-year-old Caucasian woman with Reynolds syndrome. She had a long history of Raynaud phenomenon, telangiectasia, and mild cholestasis. ... Molecular Genetics In a 76-year-old Caucasian woman with Reynolds syndrome, Gaudy-Marqueste et al. (2010) identified a heterozygous mutation in the LBR gene (R372C; 600024.0007).
SATB2 -associated syndrome is a condition that affects several body systems. ... Individuals with SATB2 -associated syndrome typically have mild to severe intellectual disability, and their ability to speak is delayed or absent. ... Causes SATB2 -associated syndrome is caused by genetic changes that affect the SATB2 gene. ... Reduction of SATB2 function likely impairs normal development of the brain and craniofacial structures, leading to intellectual disability, delayed speech, craniofacial anomalies, and other features of SATB2 -associated syndrome. The signs and symptoms of SATB2 -associated syndrome are usually similar, regardless of the type of mutation that causes it. ... Learn more about the gene and chromosome associated with SATB2-associated syndrome SATB2 chromosome 2 Inheritance Pattern SATB2 -associated syndrome is not typically inherited.
2q33.1 microdeletion syndrome is a rare chromosomal anomaly syndrome, resulting from the partial deletion of the long arm of chromosome 2, with a highly variable phenotype typically characterized by severe intellectual disability, moderate to severe developmental delay (particularly speech), feeding difficulties, failure to thrive, hypotonia, thin, sparse hair, various dental abnormalities and cleft/high-arched palate.
2q32q33 microdeletion syndrome is a recently described syndrome characterized by a variable phenotype involving moderate to severe intellectual deficit, significant speech delay, persistent feeding difficulties, growth retardation and dysmorphic features.
A number sign (#) is used with this entry because Glass syndrome (GLASS) is caused by heterozygous interstitial deletion on chromosome 2q32-q33. The disorder can also be caused by heterozygous mutation in the SATB2 gene (608148), which is within the Glass syndrome chromosome region. Description Glass syndrome is characterized by intellectual disability of variable severity and dysmorphic facial features, including micrognathia, downslanting palpebral fissures, cleft palate, and crowded teeth. ... Inheritance All patients with Glass syndrome have been shown to carry de novo heterozygous mutations in the SATB2 gene or de novo heterozygous deletions of chromosome 2q32-q33 (Leoyklang et al., 2013). ... In a 20-year-old man with Glass syndrome, Lieden et al. (2014) identified a de novo heterozygous intragenic duplication of the SATB2 gene (608148.0002). ... In a 10-year-old girl with Glass syndrome, Kaiser et al. (2015) identified a de novo heterozygous intragenic duplication of the SATB2 gene (608148.0003), predicted to result in haploinsufficiency.
Diagnostic methods Malan syndrome is a clinically recognizable overgrowth syndrome. ... Differential diagnosis The differential diagnoses include Sotos syndrome, Weaver syndrome and Marfan or Marfan-like syndromes. ... The recent implementation of prenatal whole exome sequencing could lead to molecular diagnostics during pregnancy. Genetic counseling Malan syndrome is inherited in an autosomal dominant manner. ... Management and treatment Management of Malan syndrome requires a multidisciplinary approach with appropriate medical specialists for intellectual disability, seizures, musculoskeletal and ocular abnormalities. Special education training along with behavioral intervention therapy may also be required. Prognosis Malan syndrome has significant impact on quality of life. * European Reference Network
A number sign (#) is used with this entry because Sotos syndrome-2 (SOTOS2) is caused by heterozygous mutation in the NFIX gene (164005) on chromosome 19p13. Marshall-Smith syndrome (MRSHSS; 602535) is also caused by heterozygous mutation in the NFIX gene. ... One of the patients had previously been diagnosed with a 'Sotos-like syndrome.' Inheritance Most reported cases of Sotos syndrome-2 have been sporadic and may represent new dominant mutations (Malan et al., 2010; Yoneda et al., 2012). Cytogenetics Malan et al. (2010) used a high-resolution array CGH in 18 patients with unexplained syndromic overgrowth and identified 2 patients with a de novo 19p13.1 monosomy. ... Molecular Genetics In a patient who had previously been diagnosed with a 'Sotos-like syndrome,' Malan et al. (2010) identified a heterozygous de novo nonsense mutation (Q190X; 164005.0001) in the NFIX gene.
Corneal-cerebellar syndrome Other names Der Kaloustian-Jarudi-Khoury syndrome, corneal dystrophy with spinocebellar degeneration and spinocerebellar degeneration-corneal dystrophy syndrome Corneal-cerebellar syndrome is inherited in an autosomal recessive manner Corneal-cerebellar syndrome (also known as Der Kaloustian-Jarudi-Khoury syndrome ) is an autosomally recessive disease that was first described in 1985. [1] [2] Three cases are known: all are sisters in the same family. [1] Contents 1 Symptoms 2 Cause 3 Diagnosis 3.1 Differential diagnosis 4 Treatment 5 See also 6 References 7 Further reading 8 External links Symptoms [ edit ] The age of onset is in a child's infancy . [1] Bilateral corneal opacification started in the second year of life and led to severe visual impairment. ... You can help by adding to it . ( August 2017 ) Diagnosis [ edit ] Differential diagnosis [ edit ] It was concluded by Mousa-Al et al. that the disease is different from a disease known as spastic ataxia-corneal dystrophy syndrome that had been found a year later in 1986 in an inbred Bedouin family. [2] Corneal-cerebellar syndrome differs from the spastic ataxia-corneal dystrophy syndrome by causing mental retardation. ... You can help by adding to it . ( August 2017 ) See also [ edit ] Medicine portal Rare disease References [ edit ] ^ a b c d "Orphanet: Corneal cerebellar syndrome" . Orphanet . October 2006 . Retrieved 18 May 2016 . ^ a b c d "OMIM Entry - 271310 - SPINOCEREBELLAR DEGENERATION AND CORNEAL DYSTROPHY" . ... Report of a Bedouin family—a new syndrome". J. Neurol. Sci . 76 (1): 105–21. doi : 10.1016/0022-510x(86)90145-0 .
A rare, genetic, neurological disorder characterized by the association of slowly progressive spinocerebellar degeneration and corneal dystrophy, manifesting with bilateral corneal opacities (which lead to severe visual impairment), mild intellectual disability, ataxia, gait disturbances, and tremor. Additional manifestations include facial dysmorphism (i.e. triangular face, ptosis, low-set, posteriorly angulated ears, and micrognathia), as well as mild upper motor neuron involvement with hypertonia, lower limb hyperreflexia and extensor plantar responses. There have been no further descriptions in the literature since 1985.
Clinical Features Der Kaloustian et al. (1985) reported 2 sisters born to normal but consanguineous parents with the unusual combination of spinocerebellar degeneration and corneal dystrophy. Manifestations included mental subnormality, bilateral corneal opacification starting in the second year of life and leading to severe visual impairment, and slowly progressive cerebellar abnormalities with variable dorsal column and upper motor neuron involvement. Penetrating keratoplasty resulted in improved vision. Histology of the cornea demonstrated corneal edema, thickened Descemet membrane, and degenerative pannus. Histologic abnormalities were found in muscle and sural nerve. A sister had minor spinocerebellar signs but no corneal abnormality. See 271320. Inheritance The transmission pattern of spinocerebellar degeneration and corneal dystrophy in the family described by Der Kaloustian et al. (1985) was consistent with autosomal recessive inheritance.
Perrault syndrome is a rare condition that causes different patterns of signs and symptoms in affected males and females. ... Neurological problems occur in some affected males and females. In Perrault syndrome, the problems with hearing are caused by changes in the inner ear, which is known as sensorineural hearing loss. ... Unless hearing is completely impaired at birth, the hearing problems worsen over time. Females with Perrault syndrome have abnormal or missing ovaries (ovarian dysgenesis), although their external genitalia are normal. ... However, not everyone with this condition has neurological problems. Frequency Perrault syndrome is a rare disorder; fewer than 100 affected individuals have been described in the medical literature. It is likely that the condition is underdiagnosed, because males without an affected sister will likely be misdiagnosed as having isolated (nonsyndromic) hearing loss rather than Perrault syndrome. Causes Perrault syndrome has several genetic causes.
Other features of Floating-Harbor syndrome can include an unusually high-pitched voice and, in males, undescended testes (cryptorchidism). Frequency Floating-Harbor syndrome is a rare disorder; only about 50 cases have been reported in the medical literature. Causes Floating-Harbor syndrome is caused by mutations in the SRCAP gene. ... However, the relationship between SRCAP gene mutations and the specific signs and symptoms of Floating-Harbor syndrome is unknown. Rubinstein-Taybi syndrome, a disorder with similar features, is caused by mutations in the CREBBP gene itself. ... Most cases of Floating-Harbor syndrome result from new mutations in the gene and occur in people with no history of the disorder in their family.
A multiple congenital anomalies/dysmorphic syndrome-intellectual disability that is characterized by facial dysmorphism, short stature with delayed bone age, and expressive language delay. Epidemiology Floating-Harbor syndrome prevalence and incidence are unknown. ... Associated genitourinary and cardiac anomalies have been reported in a few cases. Etiology The syndrome is associated with heterozygous mutations in exon 33 or mostly in exon 34 of the SRCAP gene (16p11.2), with two recurrent mutations (Arg2444* and Arg2435*). SRCAP encodes an ATPase which is involved in chromatin remodeling and is the cofactor of CREBBP , the gene responsible for Rubinstein-Taybi syndrome. Diagnostic methods Diagnosis is based on clinical examination and can be confirmed by genetic testing. Differential diagnosis The differential diagnosis should include other dysmorphic syndromes, in particular Rubinstein-Taybi syndrome.
Clinical Features Robinson et al. (1988) used the designation Floating-Harbor syndrome for a syndrome that was first described by Pelletier and Feingold (1973) in a boy seen at the Boston Floating Hospital and by Leisti et al. (1975) in a patient at the Harbor General Hospital in Torrance, California. ... Feingold (2006) emphasized that the correct diagnosis of the syndrome should be based on the characteristic facial features. ... Smeets et al. (1996) reported a girl with typical manifestations of the syndrome. The authors noted that marked speech delay and odd and hyperkinetic behavior may also be found in small children with Shprintzen velocardiofacial syndrome (192430). ... Celiac disease had been described in at least 3 other patients with Floating-Harbor syndrome. Ala-Mello and Peippo (1996) suggested that all patients with this syndrome should be examined for celiac disease. ... Wiltshire et al. (2005) reported a patient with Floating-Harbor syndrome complicated by a tethered spinal cord.
"The phenotype of Floating-Harbor syndrome in 10 patients". American Journal of Medical Genetics. ... "A variant example of familial Floating-Harbor syndrome?". Genetic Counseling . 14 (1): 31–7. ... (January 2013). "Not all floating-harbor syndrome cases are due to mutations in exon 34 of SRCAP". ... "The Floating-Harbor syndrome". Birth Defects Original Article Series . 11 (5): 305. ... "Search for a gene responsible for Floating-Harbor syndrome on chromosome 12q15q21.1". American Journal of Medical Genetics.
Diagnosis Suggestive Findings Floating-Harbor syndrome (FHS) should be suspected in individuals with the following clinical and radiographic features. ... Clinical Characteristics Clinical Description Prior to the molecular characterization of Floating-Harbor syndrome (FHS) by Hood et al [2012], a number of reports included descriptions of individuals in whom the diagnosis of FHS could be questioned. ... Differential Diagnosis The distinctive facial features, bone age delay, and characteristic speech disability that make the diagnosis of Floating-Harbor syndrome (FHS) straightforward in early childhood become less distinct with age. ... Other Genes of Interest in the Differential Diagnosis of Floating-Harbor Syndrome (FHS) View in own window Gene(s) 1 Disorder MOI Clinical Features of Differential Diagnosis Disorder Overlapping w/FHS Distinguishing from FHS CCDC8 CUL7 OBSL1 Three M syndrome AR Triangular face Short 5th fingers Bone age may be slightly delayed. ... Recommended Evaluations Following Initial Diagnosis in Individuals with Floating-Harbor Syndrome View in own window System/Concern Evaluation Comment Constitutional Measurement of growth & plotting of growth parameters Syndrome-specific charts are currently not available for children w/a SRCAP pathogenic variant.
Floating-Harbor syndrome (FHS) is named after the two hospitals that reported the first cases in the 1970s: Boston Floating Hospital and Harbor General Hospital in California. ... The mutation can be inherited from a parent or can occur for the first time in a person with the syndrome. Communication issues and developmental disabilities may be helped with early intervention programs and special education.
Overview Short bowel syndrome is a condition in which your body is unable to absorb enough nutrients from the foods you eat because you don't have enough small intestine. ... Symptoms Common signs and symptoms of short bowel syndrome may include: Diarrhea Greasy, foul-smelling stools Fatigue Weight loss Malnutrition Swelling (edema) in the lower extremities Causes Causes of short bowel syndrome include having parts of your small intestine removed during surgery, or being born with some of the small intestine missing or damaged. ... Diagnosis To diagnose short bowel syndrome, your doctor may recommend blood or stool tests to measure nutrient levels. ... Short bowel syndrome treatment may include: Nutritional therapy. People with small bowel syndrome will need to follow a special diet and take nutritional supplements.
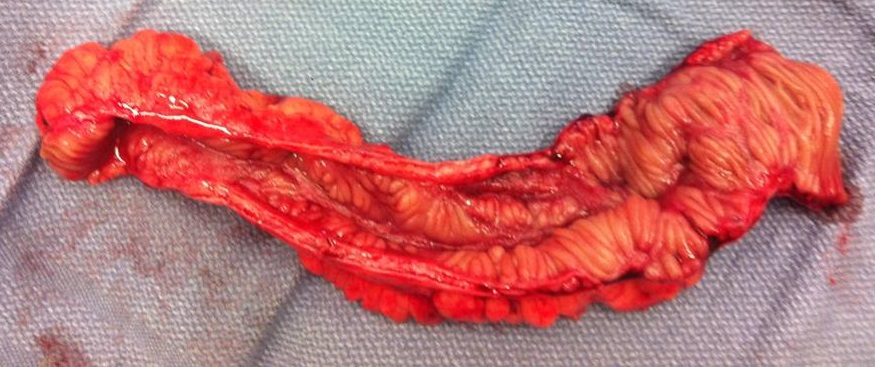
Short bowel syndrome Other names Short gut syndrome, short gut, intestinal failure A piece of diseased ileum following removal by surgery. ... See also [ edit ] Bowel-associated dermatosis–arthritis syndrome , another syndrome that can result from small-bowel bypass (or other causes) References [ edit ] ^ a b c d e f g h i j k l m n o p q r "Short Bowel Syndrome" . ... Short Bowel Syndrome: Practical Approach to Management . ... "FDA advisers back NPS's drug for short bowel syndrome" . Reuters . Archived from the original on November 11, 2012 . ... (September 2005). "Pediatric short bowel syndrome: redefining predictors of success" .
Short bowel syndrome is a disorder characterized by malabsorption of nutrients due to problems involving the small intestine. The small intestine is the tube-shaped organ between the stomach and large intestine, which includes the duodenum, jejunum, and ileum, where most food digestion and nutrient absorption take place. The causes of short bowel syndrome in adults include Crohn disease , mesenteric ischemia , radiation enteritis , or surgical removal of half or more of the small intestine to treat intestinal diseases or injuries.
Short bowel syndrome is an intestinal failure due to either a congenital defect, intestinal infarction or extensive surgical resection of the intestinal tract that results in a functional small intestine of less than 200cm in length and is characterized by diarrhea, nutrient malabsoption, bowel dilation and dysmobility.
Sézary syndrome is an aggressive form of a type of blood cancer called cutaneous T-cell lymphoma. ... A characteristic of Sézary cells is an abnormally shaped nucleus, described as cerebriform. People with Sézary syndrome develop a red, severely itchy rash (erythroderma) that covers large portions of their body. ... Although Sézary syndrome is sometimes referred to as a variant of another cutaneous T-cell lymphoma called mycosis fungoides, these two cancers are generally considered separate conditions. Frequency Sézary syndrome is a rare condition, although its prevalence is unknown. ... It is unclear whether these alterations play a role in Sézary syndrome, although the tendency to acquire chromosomal abnormalities (chromosomal instability) is a feature of many cancers.
Mycosis fungoides and Sezary syndrome are the most common cutaneous T-cell lymphomas. Sezary syndrome can arise de novo or can appear following years of chronic mycosis fungoides. ... Molecular Genetics Wang et al. (2015) presented a multiplatform genomic analysis of 37 patients with Sezary syndrome that implicates dysregulation of cell cycle checkpoint and T cell signaling. ... Da Silva Almeida et al. (2015) performed whole-exome sequencing of tumor-normal sample pairs from 25 patients with Sezary syndrome and 17 patients with other cutaneous T cell lymphomas (CTCLs). These analyses identified a distinctive pattern of somatic copy number alterations in Sezary syndrome, including highly prevalent chromosomal deletions involving the TP53, RB1 (614041), PTEN (601728), DNMT3A (602769), and CDKN1B (600778) tumor suppressors.
Mycosis fungoides is the most common form of a type of blood cancer called cutaneous T-cell lymphoma. Cutaneous T-cell lymphomas occur when certain white blood cells, called T cells , become cancerous; these cancers characteristically affect the skin, causing different types of skin lesions. Although the skin is involved, the skin cells themselves are not cancerous. Mycosis fungoides usually occurs in adults over age 50, although affected children have been identified. Mycosis fungoides may progress slowly through several stages, although not all people with the condition progress through all stages.
Lesions often initially develop on the trunk of the body in places that are rarely exposed to the sun, such as the buttocks. [2] The advanced stage of mycosis fungoides is characterized by generalized erythroderma (red rash covering most of the body) with severe pruritus (itching) and scaling. [4] The key difference between Sézary syndrome and the other stages of mycosis fungoides is the large number of cancer cells found in the blood ( leukemic disease ). [6] When mycosis fungoides develops to meet the criteria for Sézary syndrome, it is referred to as leukemic mycosis fungoides, Sézary syndrome preceded by mycosis fungoides, or secondary mycosis fungoides. [6] Cause [ edit ] The cause of mycosis fungoides is still unknown. ... (September 2019). "Mycosis fungoides and Sézary syndrome: 2019 update on diagnosis, risk-stratification, and management" . ... PMID 32632956 . ^ a b Larocca, Cecilia; Kupper, Thomas (February 2019). "Mycosis Fungoides and Sézary Syndrome: An Update" . Hematology/Oncology Clinics of North America . 33 (1): 103–120. doi : 10.1016/j.hoc.2018.09.001 . ... "How I treat mycosis fungoides and Sézary syndrome" . Blood . 114 (20): 4337–53. doi : 10.1182/blood-2009-07-202895 . ... Hwang ST, Janik JE, Jaffe ES, Wilson WH (March 2008). "Mycosis fungoides and Sézary syndrome". Lancet . 371 (9616): 945–57. doi : 10.1016/S0140-6736(08)60420-1 .
Classical mycosis fungoides is the most common type of mycosis fungoides (MF; see this term), a form of cutaneous T-cell lymphoma, and is characterized by slow progression from patches to more infiltrated plaques and eventually to tumors. Epidemiology The annual incidence of MF and its variants is estimated at between 1/350,000 and 1/110,000, with classical MF accounting for about 80-90% of MF cases. The male to female ratio is 2:1. Classical MF predominantly affects adults and the elderly (median age at diagnosis: 55-60 years). Clinical description The disease first manifests by skin lesions consisting of flat patches, preferentially located asymmetrically on the buttocks and other sun-protected areas (lower trunk and thighs, and the breasts in women). Usually, patches are hypo- or hyperpigmented in dark-skinned individuals.
Mycosis fungoides is a disease in which T-cell lymphocytes (a type of white blood cell) become malignant (cancerous) and affect the skin. This condition is one of the most common types of T-cell lymphoma . Mycosis fungoides is characterized by a scaly, red rash that develops on the skin, particularly on areas that are not usually exposed to the sun. The rash may last for months or years without causing any symptoms. Over time, a thin, reddened, eczema-like rash may develop, followed by thickened, red patches of skin. Finally, tumors form which may develop into ulcers and become infected. Mycosis fungoides is difficult to cure. Treatment is usually palliative, with the intention of relieving symptoms and improving the quality of life.
Foix–Alajouanine syndrome Other names Familial osteosclerosis with abnormalities of the nervous system and meninges Specialty Neurology Foix–Alajouanine syndrome , also called subacute ascending necrotizing myelitis , [1] is a disease caused by an arteriovenous malformation of the spinal cord . [2] The patients present with symptoms indicating spinal cord involvement ( paralysis of arms and legs, numbness and loss of sensation and sphincter dysfunction), and pathological examination reveals disseminated nerve cell death in the spinal cord and abnormally dilated and tortuous vessels situated on the surface of the spinal cord. ... See also [ edit ] Vascular myelopathy References [ edit ] ^ "Foix-Alajouanine syndrome" . Orphanet . ^ Mishra R, Kaw R (May 2005). "Foix–Alajouanine syndrome: an uncommon cause of myelopathy from an anatomic variant circulation". ... v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis v t e Focal lesions of the spinal cord General Myelopathy Myelitis Spinal cord compression By location Brown-Séquard syndrome Posterior cord syndrome Anterior cord syndrome Central cord syndrome Cauda equina syndrome Other Polio Demyelinating disease Transverse myelitis Tropical spastic paraparesis Epidural abscess Syringomyelia Syringobulbia Morvan's syndrome Sensory ataxia Tabes dorsalis Abadie's sign Subacute combined degeneration of spinal cord Vascular myelopathy Anterior spinal artery syndrome Foix–Alajouanine syndrome This article about a medical condition affecting the nervous system is a stub .
Hyaline fibromatosis syndrome is a disorder in which a clear (hyaline) substance abnormally accumulates in body tissues. ... Another common feature of hyaline fibromatosis syndrome is painful skin bumps that frequently appear on the hands, neck, scalp, ears, and nose. ... Bone abnormalities can also occur in hyaline fibromatosis syndrome. Although individuals with hyaline fibromatosis syndrome have severe physical limitations, mental development is typically normal. ... Causes Hyaline fibromatosis syndrome is caused by mutations in a gene called ANTXR2 . ... Alternatively, the mutations could impair the breakdown of excess extracellular matrix proteins, which then accumulate in tissues and lead to the signs and symptoms of hyaline fibromatosis syndrome. Researchers are unsure why the severity of hyaline fibromatosis syndrome varies among affected individuals.
A number sign (#) is used with this entry because hyaline fibromatosis syndrome (HFS) is caused by homozygous or compound heterozygous mutation in the gene encoding capillary morphogenesis protein-2 (CMG2, or ANTXR2; 608041) on chromosome 4q21. Description Hyaline fibromatosis syndrome is an autosomal recessive condition characterized by abnormal growth of hyalinized fibrous tissue usually affecting subcutaneous regions on the scalp, ears, neck, face, hands, and feet. ... Denadai et al. (2012) reported a pair of sibs and 3 other unrelated patients, all of Brazilian origin, with hyaline fibromatosis syndrome. The sibs developed pearly skin papules on the face and neck and cutaneous nodules on the ears, scalp, and fingers at ages 3 and 8 months, respectively. ... Denadai et al. (2012) concluded that it is difficult to classify patients with this disorder into an infantile or juvenile form, and suggested using the term hyaline fibromatosis syndrome (HFS), which reflects variable severity.
Juvenile hyaline fibromatosis Other names Puretic syndrome [1] Autosomal recessive pattern is the inheritance manner of this condition Specialty Dermatology Juvenile hyaline fibromatosis (also known as "Fibromatosis hyalinica multiplex juvenilis," [2] "Murray–Puretic–Drescher syndrome" [2] ) is a very rare , autosomal recessive disease due to mutations in capillary morphogenesis protein-2 (CMG-2 gene ). ... ISBN 0-07-138076-0 . ^ Casas-Alba D, Martínez-Monseny A, Pino-Ramírez RM, Alsina L, Castejón E, Navarro-Vilarrubí S, Pérez-Dueñas B, Serrano M, Palau F, García-Alix A (2018) Hyaline fibromatosis syndrome: Clinical update and phenotype-genotype correlations.
Infantile systemic hyalinosis Other names Juvenile systemic hyalinosis Infantile systemic hyalinosis is inherited in an autosomal recessive manner. Specialty Dermatology , medical genetics Infantile systemic hyalinosis is an allelic autosomal-recessive condition characterized by multiple skin nodules, hyaline deposition, gingival hypertrophy, osteolytic bone lesions and joint contractures. [1] : 606 Contents 1 Genetics 2 Diagnosis 3 Management 4 See also 5 References 6 External links Genetics [ edit ] This disease is caused by mutations in the CMG2 gene ( ANTXR2 ). [2] Diagnosis [ edit ] This section is empty. You can help by adding to it . ( October 2017 ) Management [ edit ] This section is empty. You can help by adding to it . ( October 2017 ) See also [ edit ] Skin lesion List of cutaneous conditions References [ edit ] ^ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology . (10th ed.). Saunders.
Summary Clinical characteristics. Hyaline fibromatosis syndrome (HFS) is characterized by hyaline deposits in the papillary dermis and other tissues. ... Diagnosis Suggestive Findings Hyaline fibromatosis syndrome (HFS) should be suspected in individuals with the following clinical, laboratory, histopathology, and radiographic features. ... Molecular Genetic Testing Used in Hyaline Fibromatosis Syndrome View in own window Gene 1 Method Proportion of Pathogenic Variants 2 Detectable by Method ANTXR2 Sequence analysis 3 95% 4 Gene-targeted deletion/duplication analysis 5 ~5% 6 1. ... Differential Diagnosis The conditions summarized in Table 3 exhibit some features similar to hyaline fibromatosis syndrome (HFS); however, HFS can be distinguished by the characteristic associated pain, hyperpigmented skin lesions, and perianal and perioral masses. ... MMP2 Multicentric osteolysis nodulosis & arthropathy 1 AR Short stature & osteolysis of interphalangeal & metacarpal-phalangeal joints PDGFRB Congenital generalized fibromatosis (OMIM 228550) AD Solitary, multiple, or generalized nodules composed of cells w/features of differentiated fibroblasts & smooth muscle cells AD = autosomal dominant; AR = autosomal recessive; HFS = hyaline fibromatosis syndrome; MOI = mode of inheritance 1. In addition to multicentric osteolysis nodulosis and arthropathy (MONA), this phenotype has been reported in the literature as Torg syndrome, Winchester-Torg (or Torg-Winchester) syndrome, and nodulosis-arthropathy-osteolysis (NAO) syndrome.
Differential diagnosis Juvenile hyaline fibromatosis, Winchester syndrome, lipoid proteinosis (Urbach-Wiethe disease) and mucopolysaccharidosis type II (Hunter's syndrome) should be considered in the differential diagnosis (see these terms).
Hyaline fibromatosis syndrome (HFS) is a condition characterized by deposits of a clear substance (hyaline) in the skin and in various other body tissues.
A rare genetic disease characterized by infantile or childhood onset of abnormal growth of hyalinized fibrous tissue, giving rise to multiple cutaneous nodules and/or pearly papules predominantly affecting the scalp, ears, neck, face, hands, and feet. Involvement of other organs results in gingiva hyperplasia, osteolytic bone lesions, and joint contractures. Some patients exhibit visceral involvement with intractable diarrhea, increased susceptibility to infections, and severe failure to thrive.
A rare hyaline fibromatosis syndrome characterized by papulo-nodular skin lesions (especially around the head and neck), soft tissue masses, gingival hypertrophy, joint contractures, and osteolytic bone lesions in variable degrees.
Potocki-Lupski syndrome is a condition that results from having an extra copy (duplication ) of a small piece of chromosome 17 in each cell. ... This condition is also known as 17p11.2 duplication syndrome. Infants with Potocki-Lupski syndrome may have weak muscle tone (hypotonia) and swallowing difficulties (dysphagia) that lead to feeding problems. ... About 40 percent of babies with Potocki-Lupski syndrome are born with a heart defect, which in some cases is life-threatening. ... More than 50 affected individuals have been described in the medical literature. Causes Potocki-Lupski syndrome results from a duplication of genetic material at 17p11.2. ... Most cases of Potocki-Lupski syndrome result from a new (de novo) chromosomal duplication and occur in people with no history of the disorder in their families.
Summary Clinical characteristics. Potocki-Lupski syndrome (PTLS) is characterized by cognitive, behavioral, and medical manifestations. ... Individuals with Potocki-Lupski syndrome. The facial features are not strikingly dysmorphic though common findings include micrognathia (in early childhood) and downslanting palpebral fissures. ... The phenotype of significantly larger or smaller duplications within this region may be clinically distinct from Potocki-Lupski syndrome (see Genetically Related Disorders). 4. ... Clinical Characteristics Clinical Description Potocki-Lupski syndrome (PTLS) is characterized by developmental delay, intellectual disability, behavioral disturbances, organ system involvement, and mildly dysmorphic facial features [Potocki et al 2007, Treadwell-Deering et al 2010]. ... Differential Diagnosis The differential diagnosis of Potocki-Lupski syndrome (PTLS) is broad due to the wide spectrum of findings and presence of developmental delay, learning problems, and neuropsychiatric disorders – for which the differential diagnosis is extensive.
A number sign (#) is used with this entry because the Potocki-Lupski syndrome (PTLS) is a contiguous gene syndrome caused by duplication of chromosome 17p11.2. See also Smith-Magenis syndrome (SMS; 182290), which is associated with the reciprocal deletion of chromosome 17p11.2 and shows overlapping clinical features. ... Liu et al. (2011) assembled 2 patient cohorts with reciprocal genomic disorders, deletion-associated Smith-Magenis syndrome and duplication-associated Potocki-Lupski syndrome. ... To explain this, they proposed that the probability of ectopic chromosome synapsis increases with increased LCR length, and that ectopic synapsis is a necessary precursor to ectopic crossing-over. Nomenclature Potocki-Lupski syndrome was the first predicted reciprocal microduplication syndrome described, being the homologous recombination reciprocal of the Smith-Magenis syndrome microdeletion del(17)(p11.2p11.2). Because the cytogenetic nomenclature can be cumbersome when used to refer to affected individuals, Potocki et al. (2007) proposed that the 17p11.2 microduplication syndrome be referred to by the eponym 'Potocki-Lupski syndrome' (PTLS).
Potocki–Lupski syndrome Other names 17p11.2 microduplication syndrome ,Trisomy 17p11.2 Potocki–Lupski syndrome ( PTLS ), also known as dup(17)p11.2p11.2 syndrome , trisomy 17p11.2 or duplication 17p11.2 syndrome , is a contiguous gene syndrome involving the microduplication of band 11.2 on the short arm of human chromosome 17 (17p11.2). [1] The duplication was first described as a case study in 1996. [2] In 2000, the first study of the disease was released, [3] and in 2007, enough patients had been gathered to complete a comprehensive study and give it a detailed clinical description. [1] PTLS is named for two researchers involved in the latter phases, Drs. ... Lupski of Baylor College of Medicine . [1] [4] PTLS was the first predicted reciprocal of a homologous recombination ( microdeletion or microduplication ) where both reciprocal recombinations result in a contiguous gene syndrome. [1] Its reciprocal disease is Smith–Magenis syndrome (SMS), in which the chromosome portion duplicated in PTLS is deleted altogether. [3] Potocki–Lupski syndrome is considered a rare disease , [5] [6] predicted to appear in at least 1 in 20,000 humans. [7] Symptoms of the syndrome include intellectual disability , autism , [1] and other disorders unrelated to the listed symptoms. ... Retrieved 25 August 2009 . ^ "Trisomy 17p11.2 (Potocki–Lupski syndrome)" . Orphanet . Paris, France: INSERM . Retrieved 25 August 2009 . ^ "About Potocki-Lupski syndrome" . Houston, Texas: Baylor College of Medicine . 17 July 2009. ... PMID 17024248 . ^ a b Carmona-Mora, P; Molina, J; Encina, CA; Walz, K (June 2009). "Mouse models of genomic syndromes as tools for understanding the basis of complex traits: an example with the smith-magenis and the potocki-lupski syndromes" .
Potocki-Lupski syndrome (PTLS) is a genetic disorder characterized by the presence of an extra copy of a tiny portion of chromosome 17 (duplication of 17p11.2).
17p11.2 microduplication syndrome is a rare chromosomal anomaly syndrome, resulting from the partial duplication of the short arm of chromosome 17, typically characterized by hypotonia, poor feeding, failure to thrive, developmental delay (particularly cognitive and language deficits), mild-moderate intellectual deficit, and neuropsychiatric disorders (behavioral problems, anxiety, attention deficit hyperactivity disorder, autistic spectrum disorder, bipolar disorder).
Group of delusional disorders involving belief that a person, object or place has been altered Delusional misidentification syndrome Specialty Psychiatry Delusional misidentification syndrome is an umbrella term, introduced by Christodoulou (in his book The Delusional Misidentification Syndromes , Karger, Basel, 1986) for a group of delusional disorders that occur in the context of mental and neurological illness . ... PMID 7846223 . ^ Christodoulou G.N. Delusional Misidentification Syndromes, Karger, Basel, 1986 ^ Christodoulou G.N. The Syndrome of Capgras, Br. J. Psychiatry 130, 556, 1977 ^ Christodoulou G.N. Syndrome of Subjective Doubles, Am. J. Psychiat.135,249,1978 ^ Benson DF, Gardner H, Meadows JC (February 1976). ... PMID 943070 . ^ Berrios G.E.; Luque R. (1995). "Cotard Syndrome: clinical analysis of 100 cases".
PMID 1780403 . ^ a b Josephs, K. A. (December 2007). "Capgras Syndrome and Its Relationship to Neurodegenerative Disease" . ... PMID 1961860 . ^ Sinkman A (2008). "The syndrome of capgras". Psychiatry . 71 (4): 371–378. doi : 10.1521/psyc.2008.71.4.371 . ... T.; Benson D. F. (1979). "Capgras syndrome A reduplicative phenomenon". Neurology . 29 (3): 334–9. doi : 10.1212/wnl.29.3.334 . PMID 571979 . ^ a b Barrelle, A; Luauté, JP (2018). Capgras Syndrome and Other Delusional Misidentification Syndromes . ... "Echos, Doubles, and Delusions: Capgras Syndrome in Science and Literature" . Draaisma, Douwe . 43 (3): 429.
Individuals with Wiskott-Aldrich syndrome have microthrombocytopenia, which is a decrease in the number and size of blood cells involved in clotting (platelets). ... The chance of developing certain types of cancer, such as cancer of the immune system cells (lymphoma ), is also increased in people with Wiskott-Aldrich syndrome. Wiskott-Aldrich syndrome is often considered to be part of a disease spectrum with two other disorders: X-linked thrombocytopenia and severe congenital neutropenia. ... Causes Mutations in the WAS gene cause Wiskott-Aldrich syndrome. The WAS gene provides instructions for making a protein called WASP. ... WAS gene mutations that cause Wiskott-Aldrich syndrome lead to a lack of any functional WASP. ... Learn more about the gene associated with Wiskott-Aldrich syndrome WAS Inheritance Pattern This condition is inherited in an X-linked pattern.
A number sign (#) is used with this entry because of evidence that Wiskott-Aldrich syndrome-2 (WAS2) is caused by homozygous mutation in the WIPF1 gene (602357) on chromosome 2q31. ... For a discussion of genetic heterogeneity of Wiskott-Aldrich syndrome, see WAS (301000). Clinical Features Conley et al. (1992) raised the possibility of the existence of an autosomal recessive form of Wiskott Aldrich syndrome. ... Lanzi et al. (2012) reported an 11-day-old female Moroccan infant with Wiskott-Aldrich syndrome-2. She was born to consanguineous parents who had previously lost a daughter at age 4 months with recurrent infections. The WAS2 patient presented with features of Wiskott-Aldrich syndrome, including recurrent infections, eczema, and thrombocytopenia. ... Unrelated cord blood transplantation at age 4.5 months restored immunologic function, and the WAS2 patient was healthy 16 months after the procedure. Mapping Wiskott-Aldrich syndrome-2 is caused by mutation in the WIPF1 gene, which maps to chromosome 2q31.1 (Gross, 2012).
Etiology WAS is due to hemizygous mutations in the WAS gene (Xp11.4-p11.21), coding for the Wiskott-Aldrich syndrome protein, exclusively expressed in hematopoietic cells and having a major role in the reorganization of the actin cytoskeleton, signal transduction and apoptosis.
Wiskott Aldrich syndrome (WAS) is a disease with immunological deficiency and reduced ability to form blood clots. ... Also, a skin condition known as eczema is common in people with WAS. Wiskott Aldrich syndrome is caused by mutations in the WAS gene and is inherited in an X-linked manner. ... People who have a successful and uncomplicated hematopoeitic cell transplantation, usually have normal immune function and, normal survival. Wiskott-Aldrich syndrome, X-linked thrombocytopenia (XLT), and X-linked neutropenia (XLN) are known as “ WAS -related disorders” because these diseases are all caused by mutations in the WAS gene, and have overlapping symptoms ranging from severe to mild (Wiskott-Aldrich syndrome is the most severe). The WAS gene mutations result in deficiency of the Wiskott-Aldrich syndrome protein (WASP). The more deficient the WASP, the more severe the disease.
Wiskott-Aldrich syndrome A) Multiple face petechiae and a hematoma under the right eye (left in image). ... History [ edit ] The syndrome is named after Dr. Alfred Wiskott (1898–1978), a German pediatrician who first noticed the syndrome in 1937, [42] and Dr. ... PMID 13133561 . ^ Wiskott-Aldrich Syndrome at eMedicine ^ Sande MA, Wilson WP (2001). ... "CD43, a molecule defective in Wiskott-Aldrich syndrome, binds ICAM-1". Nature . 354 (6350): 233–5. ... "Wiskott–Aldrich syndrome: diagnosis, current management, and emerging treatments" .
A number sign (#) is used with this entry because Wiskott-Aldrich syndrome (WAS) is caused by mutation in the WAS gene (300392) on chromosome Xp11. Description Wiskott-Aldrich syndrome (WAS) is an X-linked recessive immunodeficiency characterized by thrombocytopenia, eczema, and recurrent infections (Lemahieu et al., 1999). Genetic Heterogeneity of Wiskott-Aldrich Syndrome See Wiskott-Aldrich syndrome-2 (WAS2; 614493), caused by mutation in the WIPF1 gene (602357). ... Clinical Features The manifestations of Wiskott-Aldrich syndrome are eczema, thrombocytopenia, proneness to infection, and bloody diarrhea. ... They used these data 'to exclude Wiskott-Aldrich syndrome in an 18-week fetus at 50% risk of being affected.'
Neri et al. (1995) raised the possibility of an autosomal dominant form of Wiskott-Aldrich syndrome on the basis of a 3-generation family in which several members presented clinical and laboratory findings of WAS (301000), including decreased CD43 expression on T lymphocytes.
Von Hippel-Lindau syndrome is an inherited disorder characterized by the formation of tumors and fluid-filled sacs (cysts) in many different parts of the body. Tumors may be either noncancerous or cancerous and most frequently appear during young adulthood; however, the signs and symptoms of von Hippel-Lindau syndrome can occur throughout life. Tumors called hemangioblastomas are characteristic of von Hippel-Lindau syndrome. ... About 10 percent of people with von Hippel-Lindau syndrome develop endolymphatic sac tumors, which are noncancerous tumors in the inner ear . ... Frequency The incidence of von Hippel-Lindau syndrome is estimated to be 1 in 36,000 individuals. ... Most people with von Hippel-Lindau syndrome inherit an altered copy of the gene from an affected parent.
McNeill et al. (2009) proposed that patients with VHL syndrome caused by large VHL deletions that include the HSPC300 gene (C3ORF10; 611183) have a specific subtype of VHL syndrome characterized by protection from renal cell carcinoma, which the authors proposed be named VHL type 1B. ... Clinical Features The cardinal features of von Hippel-Lindau syndrome are angiomata of the retina and hemangioblastoma of the cerebellum. ... Although there were many earlier reports of this syndrome (see HISTORY), Melmon and Rosen (1964) introduced the term 'von Hippel-Landau' syndrome and described a large kindred with multiple features of the disorder. ... Griffiths et al. (1987) found reports of 6 patients with von Hippel-Lindau syndrome, pheochromocytoma, and islet cell tumor. ... These findings further supported the growing body of evidence indicating that patients with VHL syndrome caused by large VHL deletions that include the HSPC300 gene have a specific subtype of VHL syndrome with protection from RCC, which McNeill et al. (2009) proposed be named VHL type 1B.
Approximately 80% of individuals with VHL syndrome have an affected parent and about 20% have VHL syndrome as the result of a de novo pathogenic variant. ... The offspring of an individual with VHL syndrome are at a 50% risk of inheriting the VHL pathogenic variant. ... CNS hemangioblastoma is the prototypic lesion of VHL syndrome [Catapano et al 2005, Gläsker 2005]. ... Endolymphatic sac tumors are seen in approximately 10%-16% of individuals with VHL syndrome, and in some instances the associated uni- or bilateral hearing loss is the initial feature of the syndrome [Kim et al 2005, Binderup et al 2013b]. ... It is imperative to evaluate any living related potential donor for VHL syndrome and to exclude those with VHL syndrome.
Von Hippel-Lindau (VHL) disease is an inherited disorder characterized by the abnormal growth of both benign and cancerous tumors and cysts in many parts of the body. Tumors usually first appear in young adulthood. The types of tumors associated with VHL disease include hemangioblastomas (slow-growing tumors of the central nervous system); kidney cysts and clear cell renal cell carcinoma ; pancreatic neuroendocrine tumors ; pheochromocytomas (noncancerous tumors of the adrenal glands); and endolymphatic sac tumors . VHL disease is caused by a mutation in the VHL gene and is inherited in an autosomal dominant manner. Early detection and treatment of VHL disease is important, and usually involves surgical removal of tumors.
Von Hippel-Lindau disease (VHL) is a familial cancer predisposition syndrome associated with a variety of malignant and benign neoplasms, most frequently retinal, cerebellar, and spinal hemangioblastoma, renal cell carcinoma (RCC), and pheochromocytoma. ... Differential diagnosis Differential diagnoses include multiple endocrine neoplasia, neurofibromatosis, polycystic kidney disease, tuberous sclerosis, Birt-Hogg-Dube syndrome, and hereditary pheochromocytoma-paraganglioma syndromes (see these terms) associated with succinate dehydrogenase subunit mutations ( SDHB, SDHC and SDHD ).
Spider lamb syndrome , also known as spider syndrome [1] and more formally as ovine hereditary chondrodysplasia , [2] is a homozygous recessive disorder affecting the growth of cartilage and bone in sheep . ... BonDurant ^ a b c d Spider Lamb Syndrome: Introduction at UC Davis School of Veterinary Medicine ; retrieved July 19, 2012 ^ Spider Lamb Syndrome - 1998 Sheep Day Report: The Test for Spider Lamb Syndrome Gene in Sheep at North Dakota State University ; by Bert Moore, Wes Limesand and Paul Berg; publisher 1998; retrieved July 19, 2012 ^ Spider Lamb Syndrome , at the Merck Veterinary Manual ; published 2011; retrieved July 19, 2012 ^ a b Fitch, Gerald. "Spider Syndrome" (PDF) . Oklahoma Cooperative Extension Service . ^ a b "spider" . ag.ansc.purdue.edu . ... "A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome" . Am J Hum Genet . 79 (5): 935–41. doi : 10.1086/508433 . ... PMID 17033969 . ^ a b Developmental progression of the Spider Lamb Syndrome , by A.M. Oberbauer, N.E. East, R.
Aicardi-Goutières syndrome is a disorder that mainly affects the brain, the immune system, and the skin. Most newborns with Aicardi-Goutières syndrome do not show any signs or symptoms of the disorder. ... For this reason, Aicardi-Goutières syndrome is sometimes referred to as a "mimic of congenital infection." ... The encephalopathic phase of Aicardi-Goutières syndrome causes permanent neurological damage that is usually severe. ... As a result of this neurological damage, most people with Aicardi-Goutières syndrome have profound intellectual disability.
Not to be confused with Aicardi syndrome . Aicardi–Goutières syndrome Specialty Neurology , medical genetics Aicardi–Goutières syndrome ( AGS ), which is completely distinct from the similarly named Aicardi syndrome , is a rare, usually early onset childhood, inflammatory disorder most typically affecting the brain and the skin ( neurodevelopmental disorder ). [1] [2] The majority of affected individuals experience significant intellectual and physical problems, although this is not always the case. ... PMID 1641084 . ^ "Proceedings of the International Meeting on Aicardi-Goutieres Syndrome Pavia, Italy, 28-29 May 2001". ... "Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response" . ... "Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type 1 interferon signature" . ... PMID 24183309 . ^ Uggetti, C; et al. (2009). "Aicardi-Goutieres syndrome: neuroradiologic findings and follow-ups" .
Please help improve it to make it understandable to non-experts , without removing the technical details. ( June 2009 ) ( Learn how and when to remove this template message ) Conorenal syndrome Other names Mainzer-Saldino syndrome or Saldino-Mainzer disease Specialty Medical genetics Conorenal syndrome , is a collection of medical conditions that seem to have a common genetic cause. ... NPHP5 : Otto, et al. found through positional cloning that NPHP5 (IQCB1) is the most frequent cause of a disease very similar to Conorenal Syndrome (renal-retinal Senior-Loken Syndrome). ... Most diagnosis of the disease occurs when children present with kidney failure – usually between the ages of 10 and 14. There is no known cure for the syndrome and management of the syndrome is supportive. ... In 1979, Giedion [5] named the syndrome "conorenal syndrome" after a study of eight children. ... "Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin".
Mainzer-Saldino syndrome is a disorder characterized by kidney disease, eye problems, and skeletal abnormalities. People with Mainzer-Saldino syndrome have chronic kidney disease that begins in childhood and gets worse over time. ... In other people with Mainzer-Saldino syndrome, the retinal degeneration begins in childhood, but some vision is retained into early adulthood. ... However, the abnormal deposits of pigment in the retina from which retinitis pigmentosa gets its name are often not found in Mainzer-Saldino syndrome. As a result, some researchers use terms such as "atypical retinitis pigmentosa without pigment" to describe the retinal degeneration that occurs in Mainzer-Saldino syndrome. ... At least 20 cases have been reported. Causes Mainzer-Saldino syndrome is usually caused by mutations in the IFT140 gene.
SRTD encompasses Ellis-van Creveld syndrome (EVC) and the disorders previously designated as Jeune syndrome or asphyxiating thoracic dystrophy (ATD), short rib-polydactyly syndrome (SRPS), and Mainzer-Saldino syndrome (MZSDS). ... Mendley et al. (1995) extended the delineation of the conorenal syndrome to include renal histopathologic and clinical features of a primarily glomerular disorder. ... The authors suggested that this patient broadened the spectrum of Opitz trigonocephaly C syndrome with features of ciliary dysfunction. ... In addition, compound heterozygosity for mutations in IFT40 was identified in a patient with a clinical diagnosis of Jeune syndrome/ATD (614620.0002; 614620.0005). ... The findings indicated that IFT140 has a pivotal role in proper development and function of ciliated cells, and confirmed that Mainzer-Saldino syndrome is a skeletal ciliopathy. Using whole-exome sequencing, sequencing of a ciliopathy gene panel, and Sanger sequencing, Schmidts et al. (2013) screened 64 probands clinically diagnosed with Jeune syndrome/ATD and 2 with MZSDS.
Saldino-Mainzer syndrome is characterised by the association of renal disease, retinal pigmentary dystrophy, cerebellar ataxia and skeletal dysplasia. ... Femoral epiphyseal and metaphyseal anomalies are common. Genetic counseling The syndrome is transmitted as an autosomal recessive trait.