Most TBC1D24 -related disorders are inherited in an autosomal recessive manner (DOORS syndrome, FIME, PME, EIEE16, and DFNB86). ... DOORS Syndrome The five major features of DOORS syndrome are profound sensorineural hearing loss, onychodystrophy, osteodystrophy, intellectual disability / developmental delay, and seizures [James et al 2007, Campeau et al 2014]. ... Seizures, present in most individuals with DOORS syndrome, usually start in the first year of life. ... Other terms used for this condition include digito-reno-cerebral syndrome [Eronen et al 1985] and Eronen syndrome [Le Merrer et al 1992]. ... Fewer than 50 families with DOORS syndrome are known, and fewer than five each for the other TBC1D24 -related disorders.
This syndrome is sometimes referred to as the post-acute-withdrawal syndrome . ... However, trazodone is not cross-tolerant with alcohol. [12] [13] [14] The acute phase of the alcohol withdrawal syndrome can occasionally be protracted. ... In addition, patients with previous withdrawal syndromes are more likely to have more medically complicated alcohol withdrawal symptoms. ... "Outpatient management of alcohol withdrawal syndrome". American Family Physician . 88 (9): 589–95. ... "Double-blind, placebo-controlled study of the efficacy of trazodone in alcohol post-withdrawal syndrome: polysomnographic and clinical evaluations".
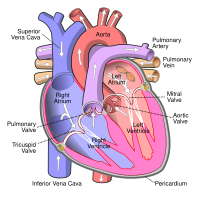
Lutembacher's syndrome This condition affects the atrium Specialty Medical genetics Lutembacher's syndrome is a very rare [1] form of congenital heart disease that affects one of the chambers of the heart (commonly the atria ) as well as a valve (commonly the mitral valve ). ... The syndrome was first described by René Lutembacher (1884–1968) [5] of Paris in 1916. [2] To correct Lutembacher's syndrome, surgery is often done. ... "Trans-Catheter Therapy of Lutembacher Syndrome: A Case Report". Acta Medica Iranica . 49 (5): 327–330. ... "Lutembacher Syndrome Workup" . ^ a b c d e f g "Atrial Septal Defect (ASD)" . ... "Percutaneous treatment of Lutembacher syndrome in a case with difficult mitral valve crossing".
A higher percentage will eventually lose the ability to walk within a ten-year interval. [11] History [ edit ] For several decades, the term tropical spastic paraparesis was used to describe a chronic and progressive clinical syndrome that affected adults living in equatorial areas of the world. This condition was initially thought to be associated with infectious agents (such as Treponema pertenue and Treponema pallidum , which cause inflammation of the central nervous system ) and with chronic nutritional deficiencies (such as avitaminosis ) or exposure to potentially toxic foods (such as bitter cassava ). [ medical citation needed ] Tropical myeloneuropathies are classified as two separate syndromes: tropical ataxic neuropathy (TAN) and tropical spastic paraparesis (TSP). ... External links [ edit ] Classification D ICD - 10 : G04.1 MeSH : D015493 DiseasesDB : 29487 External resources eMedicine : med/1038 Patient UK : Tropical spastic paraparesis Orphanet : 289326 Scholia has a topic profile for Tropical spastic paraparesis . v t e Focal lesions of the spinal cord General Myelopathy Myelitis Spinal cord compression By location Brown-Séquard syndrome Posterior cord syndrome Anterior cord syndrome Central cord syndrome Cauda equina syndrome Other Polio Demyelinating disease Transverse myelitis Tropical spastic paraparesis Epidural abscess Syringomyelia Syringobulbia Morvan's syndrome Sensory ataxia Tabes dorsalis Abadie's sign Subacute combined degeneration of spinal cord Vascular myelopathy Anterior spinal artery syndrome Foix–Alajouanine syndrome v t e Infectious diseases – viral systemic diseases Oncovirus DNA virus HBV Hepatocellular carcinoma HPV Cervical cancer Anal cancer Penile cancer Vulvar cancer Vaginal cancer Oropharyngeal cancer KSHV Kaposi's sarcoma EBV Nasopharyngeal carcinoma Burkitt's lymphoma Hodgkin lymphoma Follicular dendritic cell sarcoma Extranodal NK/T-cell lymphoma, nasal type MCPyV Merkel-cell carcinoma RNA virus HCV Hepatocellular carcinoma Splenic marginal zone lymphoma HTLV-I Adult T-cell leukemia/lymphoma Immune disorders HIV AIDS Central nervous system Encephalitis / meningitis DNA virus Human polyomavirus 2 Progressive multifocal leukoencephalopathy RNA virus MeV Subacute sclerosing panencephalitis LCV Lymphocytic choriomeningitis Arbovirus encephalitis Orthomyxoviridae (probable) Encephalitis lethargica RV Rabies Chandipura vesiculovirus Herpesviral meningitis Ramsay Hunt syndrome type 2 Myelitis Poliovirus Poliomyelitis Post-polio syndrome HTLV-I Tropical spastic paraparesis Eye Cytomegalovirus Cytomegalovirus retinitis HSV Herpes of the eye Cardiovascular CBV Pericarditis Myocarditis Respiratory system / acute viral nasopharyngitis / viral pneumonia DNA virus Epstein–Barr virus EBV infection / Infectious mononucleosis Cytomegalovirus RNA virus IV : Human coronavirus 229E / NL63 / HKU1 / OC43 Common cold MERS coronavirus Middle East respiratory syndrome SARS coronavirus Severe acute respiratory syndrome SARS coronavirus 2 Coronavirus disease 2019 V , Orthomyxoviridae : Influenza virus A / B / C / D Influenza / Avian influenza V, Paramyxoviridae : Human parainfluenza viruses Parainfluenza Human orthopneumovirus hMPV Human digestive system Pharynx / Esophagus MuV Mumps Cytomegalovirus Cytomegalovirus esophagitis Gastroenteritis / diarrhea DNA virus Adenovirus Adenovirus infection RNA virus Rotavirus Norovirus Astrovirus Coronavirus Hepatitis DNA virus HBV ( B ) RNA virus CBV HAV ( A ) HCV ( C ) HDV ( D ) HEV ( E ) HGV ( G ) Pancreatitis CBV Urogenital BK virus MuV Mumps
A tropical spastic paraparesis associated with antibody titers to human T-lymphotropic virus type 1 in the serum was described in Martinique (Gessain et al., 1985) and in Jamaica and Colombia (Rodgers-Johnson et al., 1985); a similar disorder, termed HTLV-1-associated myelopathy, has been found in parts of southwestern Japan, where adult T-cell leukemia/lymphoma (ATLL) is endemic. Osame et al. (1986) first suggested the entity of HTLV-1-associated myelopathy. Miyai et al. (1987) described 2 families with multiple cases of HTLV-1-associated myelopathy. Even though ATLL and HAM were observed in the same area of Japan, there were no observations of the disorders in the same family. It is not known whether the virus that causes ATLL is the same as that associated with HAM, although they seem to be morphologically and immunologically similar.
HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP) is a progressive disease of the nervous system that affects less than 2% of people with HTLV-1 infection . Signs and symptoms vary but may include slowly progressive weakness and spasticity of one or both legs, exaggerated reflexes, muscle contractions in the ankle, and lower back pain. Other features may include urinary incontinence and minor sensory changes, especially burning or prickling sensations and loss of vibration sense. The reason some people with HTLV-1 infection develop HAM/TSTP is not well understood. Treatment generally aims to control the specific symptoms, as there is no standard treatment available.
Tropical spastic paraparesis is a chronic systemic immune-mediated inflammatory myeloneuropathy, more frequently reported in women than in men, that usually presents in adulthood with slowly progressive spastic paraparesis of the lower limbs, bladder and bowel dysfunction, and sensory disturbances in the lower extremities (e.g. paresthesia and dysesthesia) and that is associated with a human T-cell lymphotropic virus type 1 (HTLV-1) infection.
Find sources: "Myoclonic astatic epilepsy" – news · newspapers · books · scholar · JSTOR ( March 2018 ) ( Learn how and when to remove this template message ) Myoclonic astatic epilepsy Other names Myoclonic-astatic epilepsy, myoclonic atonic epilepsy, Doose syndrome, epilepsy with myoclonic-atonic seizures, myoclonic-astatic epilepsy in early childhood Myoclonic astatic epilepsy ( MAE ), also known as myoclonic atonic epilepsy or Doose syndrome , is a generalized idiopathic epilepsy . ... This type of seizure is exclusive to MAE and is one of the defining characteristics of this syndrome. Tonic seizures: muscle stiffening or rigidity. This seizure is rare in this syndrome. Onset [ edit ] The onset of seizures is between the ages of 2 and 5 years of age. ... Some medications are harmful to those with this syndrome and can increase seizures. Diet [ edit ] The ketogenic diet mimics some of the effects of starvation, in which the body first uses up glucose and glycogen before burning stored body fat. ... "Doose syndrome (myoclonic-astatic epilepsy): 40 years of progress".
More pronounced regurgitation that is noticed through a routine physical examination is a medical sign of disease and warrants further investigation. [ medical citation needed ] If it is secondary to pulmonary hypertension it is referred to as a Graham Steell murmur . [7] Contents 1 Signs and symptoms 2 Causes 3 Pathophysiology 4 Diagnosis 5 Treatment 6 See also 7 References 8 Further reading 9 External links Signs and symptoms [ edit ] Because pulmonic regurgitation is the result of other factors in the body, any noticeable symptoms are ultimately caused by an underlying medical condition rather than the regurgitation itself. [3] However, more severe regurgitation may contribute to right ventricular enlargement by dilation, and in later stages, right heart failure . [8] A diastolic decrescendo murmur can sometimes be identified,( heard best) over the left lower sternal border. [ medical citation needed ] Causes [ edit ] Rheumatic heart disease, Among the causes of pulmonary insufficiency are: Pulmonary hypertension [1] Infective endocarditis [1] Rheumatic heart disease [1] Connective tissue disease [8] Carcinoid syndrome [8] Congestive abnormalities [9] Tetralogy of Fallot , [10] Prosthetic heart valve [11] Pathophysiology [ edit ] The pathophysiology is due to diastolic pressure variations between the pulmonary artery and right ventricle , differences are often very small, but increase regurgitation. ... Classification D ICD - 10 : I37 , Q22 ICD - 9-CM : 424.3 , 746.09 MeSH : D011665 DiseasesDB : 11014 External resources eMedicine : med/1964 Scholia has a topic profile for Pulmonary insufficiency . v t e Cardiovascular disease (heart) Ischaemic Coronary disease Coronary artery disease (CAD) Coronary artery aneurysm Spontaneous coronary artery dissection (SCAD) Coronary thrombosis Coronary vasospasm Myocardial bridge Active ischemia Angina pectoris Prinzmetal's angina Stable angina Acute coronary syndrome Myocardial infarction Unstable angina Sequelae hours Hibernating myocardium Myocardial stunning days Myocardial rupture weeks Aneurysm of heart / Ventricular aneurysm Dressler syndrome Layers Pericardium Pericarditis Acute Chronic / Constrictive Pericardial effusion Cardiac tamponade Hemopericardium Myocardium Myocarditis Chagas disease Cardiomyopathy Dilated Alcoholic Hypertrophic Tachycardia-induced Restrictive Loeffler endocarditis Cardiac amyloidosis Endocardial fibroelastosis Arrhythmogenic right ventricular dysplasia Endocardium / valves Endocarditis infective endocarditis Subacute bacterial endocarditis non-infective endocarditis Libman–Sacks endocarditis Nonbacterial thrombotic endocarditis Valves mitral regurgitation prolapse stenosis aortic stenosis insufficiency tricuspid stenosis insufficiency pulmonary stenosis insufficiency Conduction / arrhythmia Bradycardia Sinus bradycardia Sick sinus syndrome Heart block : Sinoatrial AV 1° 2° 3° Intraventricular Bundle branch block Right Left Left anterior fascicle Left posterior fascicle Bifascicular Trifascicular Adams–Stokes syndrome Tachycardia ( paroxysmal and sinus ) Supraventricular Atrial Multifocal Junctional AV nodal reentrant Junctional ectopic Ventricular Accelerated idioventricular rhythm Catecholaminergic polymorphic Torsades de pointes Premature contraction Atrial Junctional Ventricular Pre-excitation syndrome Lown–Ganong–Levine Wolff–Parkinson–White Flutter / fibrillation Atrial flutter Ventricular flutter Atrial fibrillation Familial Ventricular fibrillation Pacemaker Ectopic pacemaker / Ectopic beat Multifocal atrial tachycardia Pacemaker syndrome Parasystole Wandering atrial pacemaker Long QT syndrome Andersen–Tawil Jervell and Lange-Nielsen Romano–Ward Cardiac arrest Sudden cardiac death Asystole Pulseless electrical activity Sinoatrial arrest Other / ungrouped hexaxial reference system Right axis deviation Left axis deviation QT Short QT syndrome T T wave alternans ST Osborn wave ST elevation ST depression Strain pattern Cardiomegaly Ventricular hypertrophy Left Right / Cor pulmonale Atrial enlargement Left Right Athletic heart syndrome Other Cardiac fibrosis Heart failure Diastolic heart failure Cardiac asthma Rheumatic fever v t e Congenital heart defects Heart septal defect Aortopulmonary septal defect Double outlet right ventricle Taussig–Bing syndrome Transposition of the great vessels dextro levo Persistent truncus arteriosus Aortopulmonary window Atrial septal defect Sinus venosus atrial septal defect Lutembacher's syndrome Ventricular septal defect Tetralogy of Fallot Atrioventricular septal defect Ostium primum Consequences Cardiac shunt Cyanotic heart disease Eisenmenger syndrome Valvular heart disease Right pulmonary valves stenosis insufficiency absence tricuspid valves stenosis atresia Ebstein's anomaly Left aortic valves stenosis insufficiency bicuspid mitral valves stenosis regurgitation Other Underdeveloped heart chambers right left Uhl anomaly Dextrocardia Levocardia Cor triatriatum Crisscross heart Brugada syndrome Coronary artery anomaly Anomalous aortic origin of a coronary artery Ventricular inversion
EZH2- related overgrowth includes EZH2- related Weaver syndrome at one end of the spectrum and tall stature at the other. Although most individuals diagnosed with a heterozygous EZH2 pathogenic variant have been identified because of a clinical suspicion of Weaver syndrome, a minority have been identified through molecular genetic testing of family members of probands or individuals with overgrowth who did not have a clinical diagnosis of Weaver syndrome. ... Although pathogenic variants in NSD1 (the cause of Sotos syndrome) were once reported to cause Weaver syndrome [Douglas et al 2003], this association has been refuted [Tatton-Brown et al 2005]. Prevalence Because EZH2 pathogenic variants have only recently been shown to cause Weaver syndrome, and individuals with a mild phenotype may escape clinical diagnosis, it is currently difficult to estimate the prevalence of Weaver syndrome. ... Pathogenic variants in NSD1 (the cause of Sotos syndrome) were once reported to cause Weaver syndrome [Douglas et al 2003].
Burkitt lymphoma (BL) is a very fast-growing type of cancer. It is a form of B-cell non-Hodgkin's lymphoma . There are 3 recognized forms of BL: Endemic (African) - the most common form, found mainly in central Africa, where it is associated with the Epstein Barr virus (EBV) . It is most common in children. This form often manifests as enlargement of the jaw or facial bones. Sporadic - a rarer form, seen in all parts of the world, that often develops in the abdomen with bone marrow involvement. The kidneys, ovaries, breasts or other organs may also be involved. This form commonly affects children and young adults.
A number sign (#) is used with this entry because of evidence that Burkitt lymphoma can be caused by somatic mutation in the MYC gene (190080) in addition to translocations involving the MYC gene and immunoglobulin genes (see 147220). Description Burkitt lymphoma is a rare, aggressive B-cell lymphoma that accounts for 30 to 50% of lymphomas in children but only 1 to 2% of lymphomas in adults (Harris and Horning, 2006). It results from chromosomal translocations that involve the MYC gene (190080) and either the lambda or the kappa light chain immunoglobulin genes (147220, 147200). Burkitt lymphoma is causally related to the Epstein-Barr virus (EBV), although the pathogenetic mechanisms are not clear. Clinical Features Anderson et al. (1986) described 2 sisters in an American family who died of Burkitt lymphoma at ages 11 and 22 years.
A number sign (#) is used with this entry because of evidence that susceptibility to acute lymphoblastic leukemia-3 (ALL3) is conferred by heterozygous mutation in the PAX5 gene (167414) on chromosome 9p13. For a general phenotypic description and a discussion of genetic heterogeneity of acute lymphoblastic leukemia, see 613065. Clinical Features Shah et al. (2013) reported 2 unrelated families in which multiple individuals in several generations had childhood onset of B-cell acute lymphoblastic leukemia (B-ALL). Relapse was common. One of the families was of Puerto Rican descent and the other was of African American descent. Gu et al. (2019) characterized 2 subtypes of B-ALL, PAX5alt (PAX5-altered) and PAX5 pro80 to arg (P80R), that are defined by PAX5 alterations (see MOLECULAR GENETICS).
Burkitt lymphoma is a rare form of malignant mature B-cell non-Hodgkin lymphoma. Epidemiology In Europe and north America it represents around half of all malignant non-Hodgkin lymphoma in children and around 2% in adults. Two incidence peaks occur: one in childhood/adolescence and the second after the age of 40 years. In Europe the standardized incidence ratio is 1/530,000 for individuals aged between 0 and 14 years and 1/670,000 for individuals aged between 15 and 19 years. Males are affected more than females. Clinical description Patients with HIV in whom antiviral treatment is ineffective are particularly susceptible to Burkitt lymphoma.
Chronic granulomatous disease Other names Bridges–Good syndrome, chronic granulomatous disorder, Quie syndrome Superoxide Specialty Immunology Chronic granulomatous disease ( CGD ), also known as Bridges–Good syndrome , chronic granulomatous disorder , and Quie syndrome , [1] is a diverse group of hereditary diseases in which certain cells of the immune system have difficulty forming the reactive oxygen compounds (most importantly the superoxide radical due to defective phagocyte NADPH oxidase ) used to kill certain ingested pathogens . [2] This leads to the formation of granulomas in many organs. [3] CGD affects about 1 in 200,000 people in the United States , with about 20 new cases diagnosed each year. [4] [5] This condition was first discovered in 1950 in a series of 4 boys from Minnesota, and in 1957 it was named "a fatal granulomatosus of childhood" in a publication describing their disease. [6] [7] The underlying cellular mechanism that causes chronic granulomatous disease was discovered in 1967, and research since that time has further elucidated the molecular mechanisms underlying the disease. [8] Bernard Babior made key contributions in linking the defect of superoxide production of white blood cells, to the cause of the disease. ... Small groups of CGD patients may also be affected by McLeod syndrome because of the proximity of the two genes on the same X-chromosome. [ citation needed ] Atypical infections [ edit ] Microscopic image of the fungus, Aspergillus fumigatus , an organism that commonly causes disease in people with chronic granulomatous disease. ... "A fatal granulomatosus of childhood: the clinical study of a new syndrome". Minnesota Medicine . 40 (5): 309–12. ... "A fatal granulomatous disease of childhood; the clinical, pathological, and laboratory features of a new syndrome". A.M.A. Journal of Diseases of Children . 97 (4): 387–408. doi : 10.1001/archpedi.1959.02070010389004 . ... Retrieved 2019-10-22 . v t e Diseases of monocytes and granulocytes Monocytes and macrophages ↑ -cytosis : Monocytosis Histiocytosis Chronic granulomatous disease ↓ -penia : Monocytopenia Granulocytes ↑ -cytosis : granulocytosis Neutrophilia Eosinophilia / Hypereosinophilic syndrome Basophilia Bandemia ↓ -penia : Granulocytopenia/agranulocytosis ( Neutropenia / Severe congenital neutropenia / Cyclic neutropenia Eosinopenia Basopenia ) Disorder of phagocytosis Chemotaxis and degranulation Leukocyte adhesion deficiency LAD1 LAD2 Chédiak–Higashi syndrome Neutrophil-specific granule deficiency Respiratory burst Chronic granulomatous disease Neutrophil immunodeficiency syndrome Myeloperoxidase deficiency v t e X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) Wiskott–Aldrich syndrome X-linked severe combined immunodeficiency X-linked agammaglobulinemia Hyper-IgM syndrome type 1 IPEX X-linked lymphoproliferative disease Properdin deficiency Hematologic Haemophilia A Haemophilia B X-linked sideroblastic anemia Endocrine Androgen insensitivity syndrome / Spinal and bulbar muscular atrophy KAL1 Kallmann syndrome X-linked adrenal hypoplasia congenita Metabolic Amino acid : Ornithine transcarbamylase deficiency Oculocerebrorenal syndrome Dyslipidemia : Adrenoleukodystrophy Carbohydrate metabolism : Glucose-6-phosphate dehydrogenase deficiency Pyruvate dehydrogenase deficiency Danon disease/glycogen storage disease Type IIb Lipid storage disorder : Fabry's disease Mucopolysaccharidosis : Hunter syndrome Purine–pyrimidine metabolism : Lesch–Nyhan syndrome Mineral : Menkes disease / Occipital horn syndrome Nervous system X-linked intellectual disability : Coffin–Lowry syndrome MASA syndrome Alpha-thalassemia mental retardation syndrome Siderius X-linked mental retardation syndrome Eye disorders: Color blindness (red and green, but not blue) Ocular albinism ( 1 ) Norrie disease Choroideremia Other: Charcot–Marie–Tooth disease (CMTX2-3) Pelizaeus–Merzbacher disease SMAX2 Skin and related tissue Dyskeratosis congenita Hypohidrotic ectodermal dysplasia (EDA) X-linked ichthyosis X-linked endothelial corneal dystrophy Neuromuscular Becker's muscular dystrophy / Duchenne Centronuclear myopathy (MTM1) Conradi–Hünermann syndrome Emery–Dreifuss muscular dystrophy 1 Urologic Alport syndrome Dent's disease X-linked nephrogenic diabetes insipidus Bone / tooth AMELX Amelogenesis imperfecta No primary system Barth syndrome McLeod syndrome Smith–Fineman–Myers syndrome Simpson–Golabi–Behmel syndrome Mohr–Tranebjærg syndrome Nasodigitoacoustic syndrome X-linked dominant X-linked hypophosphatemia Focal dermal hypoplasia Fragile X syndrome Aicardi syndrome Incontinentia pigmenti Rett syndrome CHILD syndrome Lujan–Fryns syndrome Orofaciodigital syndrome 1 Craniofrontonasal dysplasia
Some males with X-linked CGD have McLeod neuroacanthocytosis syndrome as the result of a contiguous gene deletion. ... Prolonged and dysregulated inflammation in CGD can overlap clinically with the syndrome of hemophagocytic lymphohistiocytosis (HLH). ... Individuals with CGD are prone to recurrent infection with different strains of Burkholderia cepacia complex , whereas those with cystic fibrosis are often persistently infected with the same strain [Greenberg et al 2009]. Hyper IgE syndrome . Staphylococcal and Aspergillus infections are common in individuals with autosomal dominant STAT3-deficient hyper IgE syndrome (AD-HIES). ... Because the HLH-like syndrome in CGD represents a reaction to bacterial or fungal infection, these infections must be aggressively treated if patients receive immunosuppression for HLH. ... Persons with McLeod neuroacanthocytosis syndrome do not express the erythrocyte blood group Kell antigen (i.e., they are Kell negative).
Berendes et al. (1957) and Bridges et al. (1959) identified a new syndrome which they termed 'fatal granulomatosis of childhood.' ... Carson et al. (1965) reported 16 males in 8 families with a syndrome of chronic suppurative lymphadenitis, chronic dermatitis, chronic pulmonary disease and hepatosplenomegaly with subsequent fatal outcome. ... Segal (1985) gave a useful review of the molecular basis of CGD, viewed as a syndrome caused by any defect in the function of the electron transport chain essential to the microbicidal activity of white cells. ... One patient had X-CDG in combination with McLeod syndrome (300842). Cytogenetics Kumatori et al. (1998) concluded that nonhomologous recombination between the CYBB gene and a LINE-1 element lies 5-kb upstream of CYBB in normal persons. ... Although it appears that the coexistence of CGD and the McLeod syndrome in some patients is due to the deletion of 2 very closely linked genes, Xk and CGD, Branch et al. (1986) showed that granulocytes lack red cell Kx antigen.
A number sign (#) is used with this entry because of evidence that autosomal recessive cytochrome b-positive chronic granulomatous disease (CGD) type III is caused by compound heterozygous mutation in the NCF4 gene (601488), which encodes the p40-phox (phagocyte oxidase) protein, on chromosome 22q12. One such patient has been reported. Description Autosomal recessive cytochrome b-positive chronic granulomatous disease (CGD) type III is a immunodeficiency disorder characterized by recurrent pyogenic infections and granulomatous inflammation resulting from loss of phagocyte superoxide production (summary by Matute et al., 2009). For a general phenotypic description and a discussion of genetic heterogeneity of chronic granulomatous disease, see the well-established X-linked recessive cytochrome b-negative form (CGD; 306400). Clinical Features Matute et al. (2009) reported a 3.5-year-old boy who presented with diarrhea, low-grade fever, and perianal rash. He also had perioral eczema and aphthous ulcers. He was found to have chronic granulomatous colitis, with erosions and ulceration of the gastric fundus and colonic mucosa, and multiple small granulomata on colonic biopsy.
A number sign (#) is used with this entry because autosomal recessive cytochrome b-positive chronic granulomatous disease (CGD) type II is caused by homozygous or compound heterozygous mutation in the NCF2 gene (608515), which encodes the p67-phox (phagocyte oxidase) protein, on chromosome 1q25. A more common form of autosomal cytochrome b-positive chronic granulomatous disease, type I (233700), is caused by mutation in the NCF1 gene (608512), which encodes the p47-phox protein. For a phenotypic description of chronic granulomatous disease, see the well-established X-linked recessive cytochrome b-negative form (CGD; 306400). Clinical Features Nunoi et al. (1995) reported a Japanese patient with p67-deficient CGD confirmed by mutation in the NCF2 gene (608515.0001). He was a 19-year-old man whose first episode of infection was at age 3 when he had perianal abscess, liver abscess, severe lung abscess, pneumonia with Aspergillus infection, and severe spinal Aspergillus osteomyelitis.
Overview Chronic granulomatous (gran-u-LOM-uh-tus) disease (CGD) is an inherited disorder that occurs when a type of white blood cell, called a phagocyte, doesn't work properly. Phagocytes usually help your body fight infections. When they don't work as they should, phagocytes can't protect your body from bacterial and fungal infections. People with chronic granulomatous disease may develop infections in their lungs, skin, lymph nodes, liver, stomach and intestines, or other areas. They also may develop clusters of white blood cells in infected areas. Most people are diagnosed with CGD during childhood, but some people may not be diagnosed until adulthood.
A number sign (#) is used with this entry because autosomal recessive cytochrome b-negative chronic granulomatous disease (CGD4) is caused by mutation in the gene encoding p22-phox (CYBA; 608508). For a detailed phenotypic description of chronic granulomatous disease, see X-linked cytochrome b-negative CGD (306400). See also autosomal recessive cytochrome b-positive CGD, types I (233700) II (608515), and III (613960). Description Chronic granulomatous disease is a genetically heterogeneous immunodeficiency disorder resulting from an inability of phagocytes to kill microbes that they have ingested. This impairment in killing is caused by any of several defects in the NADPH oxidase enzyme complex which generates the microbicidal 'respiratory burst.'
Chronic granulomatous disease (CGD) is a rare, inherited immunodeficiency that affects certain white blood cells. People with this condition have immune systems that do not function properly, leaving the body vulnerable to chronic inflammation and frequent bacterial and fungal infections. The features of this condition usually develop in infancy or early childhood; however, milder forms may be diagnosed in the teen years or even in adulthood. It is caused by changes (mutations) in any one of five different genes and is usually inherited in an autosomal recessive or X-linked recessive manner. Treatment consists of continuous therapy with antibiotic and antifungal medications to treat and prevent infections.
Segal (1985) gave a useful review of the molecular basis of CGD, viewed as a syndrome caused by any defect in the function of the electron transport chain essential to the microbicidal activity of white cells. ... Kimpen et al. (1991) proposed that the autosomal recessive form of CGD might be due to a mutation on 18q because they observed the association of CGD with 18q- syndrome. In fact, the 3 forms of autosomal recessive CGD are known to map to chromosomes 1, 7, and 16.
Autoimmune disorders such as discoid lupus erythematosus and antiphospholipid syndrome can occur in some. Etiology CGD is caused by mutations in any one of the 6 genes encoding the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits or a critical stabilizer. ... Differential diagnosis Differential diagnosis includes cystic fibrosis, Crohn disease, hyper-IgE syndrome, allergic bronchopulmonary aspergillosis, glutathione synthetase deficiency, and secondary hemophagocytic lymphohistiocytosis.
Clark and Klebanoff (1978) described a brother and sister, aged 24 and 20, respectively, with recurrent staphylococcal infections with predominantly cutaneous involvement. Neutrophils showed normal phagocytosis but impaired killing of staphylococci and absence of a phagocytic metabolic burst as assessed by eight functions. The mother's neutrophils functioned normally. Both patients showed, unexpectedly, marked impairment of chemotactic responses of their neutrophils and in the level of chemotactic activity generated in their serum by activation of the complement system. Furthermore, their serum contained an inhibitor of chemotactic response by normal neutrophils. Tauber (1981) gave a useful analysis of neutrophil dysfunction, dividing disorders into those of each of the 4 behaviors or functions of the neutrophil: chemotaxis, phagocytosis, degranulation, and oxidative metabolism.
Baraitser et al. (1983) described brother and sister with distal arthrogryposis presenting as windmill-vane hand, facial immobility, and generalized ichthyosis.
A disorder defining by the association of Perineal hemangioma, External genitalia malformations, Lipomyelomeningocele, Vesicorenal abnormalities, Imperforate anus, and Skin tag. Eleven cases have been reported.
Lambert et al. (2001) suggested that karyotyping of skin fibroblasts should be performed when the diagnosis of Camera-Marugo-Cohen syndrome is considered.
Mapping By linkage studies in a family with a syndromic form of mental retardation, Abidi et al. (1999) obtained a lod score of 4.41 with zero recombination at locus DXS1166 in Xq13.2.
X-linked intellectual disability, Abidi type is characterized by X-linked intellectual deficit and mild variable manifestations, including short stature, small head circumference, sloping forehead, hearing loss, abnormally shaped ears, and small testes. It has been described in eight affected males from three generations.
Diaphragmatic defect-limb deficiency-skull defect syndrome is characterized by the association of classical diaphragmatic hernia (Bochdalek type) with severe lung hypoplasia, and variable associated malformations.
Clinical Features Froster et al. (1996) described a 'possibly new' autosomal recessive syndrome in 4 successive fetuses (2 females; 2 males) from a healthy nonconsanguineous couple. ... The specific spectrum of anomalies was not fully compatible with that of any established syndrome. No prenatal exposure to any possible teratogen was found. ... The authors proposed that the infant had the same condition as the sibs reported by Froster et al. (1996) and suggested that the condition be called Froster syndrome. Gardham et al. (2013) reported 2 sibs, born to healthy nonconsanguineous parents, with features similar to those in the patients reported by Froster et al. (1996) and Koifman et al. (2009).