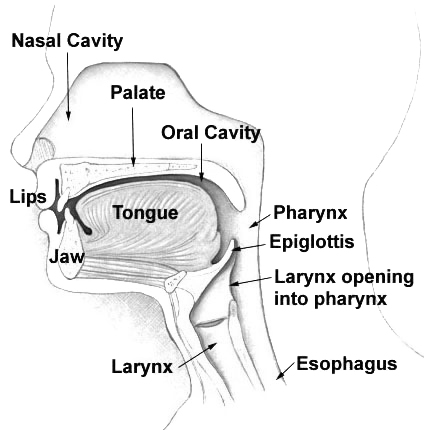
See also HLS2 (614120), caused by mutation in the KIF7 gene (611254) on chromosome 15q26. Clinical Features This lethal syndrome was discovered in Finland in the course of studying the Meckel syndrome (see 249000), which is frequent there (Salonen et al., 1981). Like the Meckel syndrome, this disorder is characterized by polydactyly and central nervous system malformation, but unlike that syndrome, it does not show cystic kidney and liver and the CNS derangement is hydrocephalus not encephalocele. ... Salonen et al. (1981) described the syndrome in 28 newborns in 18 Finnish families. ... The authors considered this case to be a variant of hydrolethalus syndrome, although absence of hydrocephaly and preaxial polydactyly of the feet may suggest other diagnoses, e.g., Joubert syndrome (213300). ... Mapping Visapaa et al. (1999) assigned the hydrolethalus syndrome locus to 11q23-q25 in Finnish families.
Hydrolethalus (HLS) is a severe fetal malformation syndrome characterized by craniofacial dysmorphic features, central nervous system, cardiac, respiratory tract and limb abnormalities. ... Differential diagnosis Differential diagnosis of HLS includes, in the absence of an index case, other midline multiple malformation syndromes like Pallister-Hall, pseudo-trisomy 13, oro-facio-digital type IV or VI, Joubert syndromes, as well as the severe form of Smith-Lemli-Opitz syndrome (see these terms). ... Genetic counseling As an autosomal recessive syndrome, HLS has a 25% recurrence risk after an affected pregnancy.
WikiProject Genetics may be able to help recruit an expert. ( August 2008 ) Hydrolethalus syndrome Other names Salonen-Herva-Norio syndrome Hydrolethalus syndrome is inherited in an autosomal recessive manner Specialty Medical genetics Hydrolethalus syndrome ( HLS ) is a rare genetic disorder that causes improper fetal development , resulting in birth defects and, most commonly, stillbirth . [1] HLS is associated with HYLS1 mutations. ... "A novel HYLS1 homozygous mutation in living siblings with Joubert syndrome". Clinical Genetics . 89 (6): 739–43. doi : 10.1111/cge.12752 . ... "KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes" . Nature Genetics . 43 (6): 601–6. doi : 10.1038/ng.826 . ... PMID 26472341 . ^ a b c d e f Salonen R, Herva R (December 1990). "Hydrolethalus syndrome" . Journal of Medical Genetics . 27 (12): 756–59. doi : 10.1136/jmg.27.12.756 . ... "Unraveling the disease pathogenesis behind lethal Hydrolethalus syndrome revealed multiple changes in molecular and cellular level" .
A number sign (#) is used with this entry because of evidence that hydrolethalus syndrome-2 (HLS2) is caused by homozygous mutation in the KIF7 gene (611254) on chromosome 15q26. One such family has been reported. Description Hydrolethalus syndrome is an autosomal recessive embryonic lethal disorder characterized by hydrocephaly or anencephaly, postaxial polydactyly of the upper limbs, and pre- or postaxial polydactyly of the lower limbs. ... HLS2 is considered a ciliopathy (summary by Putoux et al., 2011). Acrocallosal syndrome (ACLS; 200990) is an allelic disorder with a less severe phenotype. For a discussion of genetic heterogeneity of hydrolethalus syndrome, see 236680. Clinical Features Putoux et al. (2011) reported a consanguineous Algerian family in which 4 sib fetuses had a lethal developmental disorder consistent with hydrolethalus syndrome. ... Molecular Genetics By genomewide linkage analysis followed by candidate gene analysis of a consanguineous Algerian family with hydrolethalus syndrome, Putoux et al. (2011) identified a homozygous deletion in the KIF7 gene (611254.0001) in affected individuals.
Jeavons syndrome is a type of epilepsy . It is one of the most distinctive reflex syndromes of idiopathic generalized epilepsy characterized by the triad of eyelid myoclonia with and without absences , eye-closure-induced seizures, EEG paroxysms, or both, and photosensitivity . ... This is probably due to the enhancing effect of bright light on the sensitivity of eye closure. Self-induction in Jeavons syndrome [ edit ] Self-induced seizures in Jeavons syndrome are rare. ... Simply closing the eyes would be more potent. Cause [ edit ] Jeavons syndrome is a genetically determined homogeneous syndrome, with a high prevalence of similar seizures in family members. ... Reflex seizures and related epileptic syndromes. In: A Clinical Guide to Epileptic Syndromes and Their Treatment. ... London: Springer, 2010:497–531. ^ Covanis A. Jeavons syndrome. In: Panayiotopoulos CP, editor.
Epidemiology Prevalence is unknown but Jeavons syndrome appears to represent around 7-8% of all idiophatic generalized epilepsies (IGEs). The syndrome is slightly more frequent in females than in males. ... Etiology The etiology is unknown but Jeavons syndrome appears to be genetically determined: the majority of reported patients have a family history of IGE and a few cases of affected twins have been reported. Diagnostic methods Eyelid myoclonia is a highly distinctive seizure type and is strongly suggestive of Jeavons syndrome. Video-electroencephalography (video-EEG) is the only procedure required for diagnosis and reveals eye closure-related generalized paroxysmal activity. ... Prognosis The overall prognosis is good, although Jeavons syndrome is usually a lifelong condition.
This symptomatic triad (moniliform hair, follicular hyperkeratosis and nail dystrophy), when associated with other ectodermal defects, such as as neurological, dental, and ophthalmological alterations, constitute the ''moniliform hair syndrome''. Etiology Four genes have been associated with monilethrix: KRT81 , KRT83 and KRT86 , coding for the type II hair keratins Hb1, Hb3 and Hb6, and are responsible for the autosomal dominant form of the disease.
A number sign (#) is used with this entry because of evidence that monilethrix is caused by heterozygous mutation in the hair cortex keratin genes KRTHB1 (KRT81; 602153), KRTHB6 (KRT86; 601928), and KRTHB3 (KRT83; 602765). Description Individuals with monilethrix have normal hair at birth, but within the first few months of life develop fragile, brittle hair that tends to fracture and produce varying degrees of dystrophic alopecia. In the mildest forms, only the occipital regions of the scalp are involved; however, in severe forms the eyebrows, eyelashes, and secondary sexual hair may also be involved. Follicular hyperkeratosis with predilection for the scalp, nape of neck, and extensor surfaces of the upper arm and thighs is also a characteristic finding in these patients. Light microscopic examination is diagnostic and reveals elliptical nodes of normal thickness and intermittent constrictions (internodes) at which the hair easily breaks.
Monilethrix is a condition that affects hair growth. Its most characteristic feature is that individual strands of hair have a beaded appearance like the beads of a necklace. The name monilethrix comes from the Latin word for necklace (monile) and the Greek word for hair (thrix). Noticeable when viewed under a microscope, the beaded appearance is due to periodic narrowing of the hair shaft. People with monilethrix also have sparse hair growth (hypotrichosis) and short, brittle hair that breaks easily. Affected individuals usually have normal hair at birth, but the hair abnormalities develop within the first few months of life.
Monilethrix is a rare condition caused by a defect in the hair shaft resulting in hair which appears dry, dull, and brittle, and which breaks spontaneously or with mild trauma. The age of onset, severity, and course may vary from person to person.
A number sign (#) is used with this entry because hypotonia-cystinuria syndrome is a contiguous gene syndrome caused by a homozygous deletion on chromosome 2p21 that disrupts the SLC3A1 (104614) and PREPL (609557) genes. ... Larger homozygous deletions in this region, including a 179-kb deletion, result in a more severe phenotype termed the '2p21 deletion syndrome.' Genes deleted in the 2p21 deletion syndrome include SLC3A1, PREPL, PPM1B (603770), C2ORF34 (609559), and possibly other genes. ... Jaeken et al. (2006) reported 11 patients from 9 families with hypotonia-cystinuria syndrome. Seven families were Flemish and 2 were French. ... Chabrol et al. (2008) reported a brother and sister, born of unrelated Moroccan parents from the same village, with a phenotype that was intermediate between hypotonia-cystinuria syndrome and the 2p21 deletion syndrome. ... The former title 'homozygous 2p16 deletion syndrome' is retained here for historical purposes.
A form of hypotonia-cystinuria type 1 syndrome characterized by mild to moderate intellectual disability in addition to classic hypotonia-cystinuria syndrome phenotype (cystinuria type 1, generalised hypotonia, poor feeding, growth retardation, and minor facial dysmorphism).
A rare, genetic disorder of amino acid absorption and transport, characterized by generalized hypotonia at birth, neonatal/infantile failure to thrive (followed by hyperphagia and rapid weight gain in late childhood), cystinuria type 1, nephrolithiasis, growth retardation due to growth hormone deficiency, and minor facial dysmorphism. Dysmorphic features mainly include dolichocephaly and ptosis. Nephrolithiasis occurs at variable ages.
The 2p21 microdeletion syndrome consists of cystinuria, neonatal seizures, hypotonia, severe growthand developmental delay, facial dysmorphism, and lactic acidemia. ... Reduced activity of the respiratory chain complexes I, III, IV and V was found in patients examined. Etiology The syndrome is caused by homozygous deletion of at least four contiguous genes on chromosome 2: SLC3A1 , PREPL , PPM1B and C2orf34 (2p21).
Hernandez et al. (1996) described an 8-year-old boy with generalized osteoporosis and oculocutaneous hypopigmentation syndrome (OOCH) without cerebral defects. ... Psychomotor development was normal. Differences from the Cross syndrome (257800) and Preus syndrome (257790) were the lack of cerebral abnormality and the presence of osteoporosis. Skel - Generalized osteoporosis Eyes - Oculocutaneous hypopigmentation syndrome (OOCH) Neuro - No cerebral defects Inheritance - ?
Persistent adrenarche syndrome Other names Adrenal SAHA syndrome [1] Specialty Dermatology Persistent adrenarche syndrome is a cutaneous condition seen typically in thin young women who report great psychological and physical stress in their lives. [1] See also [ edit ] SAHA syndrome List of cutaneous conditions References [ edit ] ^ a b Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007).
Ring chromosome 18 syndrome is an autosomal anomaly characterized by variable clinical features, most commonly including hypotonia, neonatal feeding and respiratory difficulties, microcephaly, global developmental delay and intellectual disability, growth hormone deficiency, hypothyroidism, hearing loss, aural atresia, dysmorphic facial features and behavioral characteristics.
Infants with female sex assignment present with varying degrees of virilization and may show manifestations of other clinical features of Turner syndrome (see this term). The uterus is of variable size and the degree of differentiation of the internal genitalia varies. ... Differential diagnosis The differential diagnosis should include 46,XY partial gonadal dysgenesis (46,XY PGD; see this term) and syndromic 46,XY gonadal dysgenesis (such as Frasier syndrome, campomelic dysplasia and 46,XY DSD with adrenal insufficiency; see these terms).
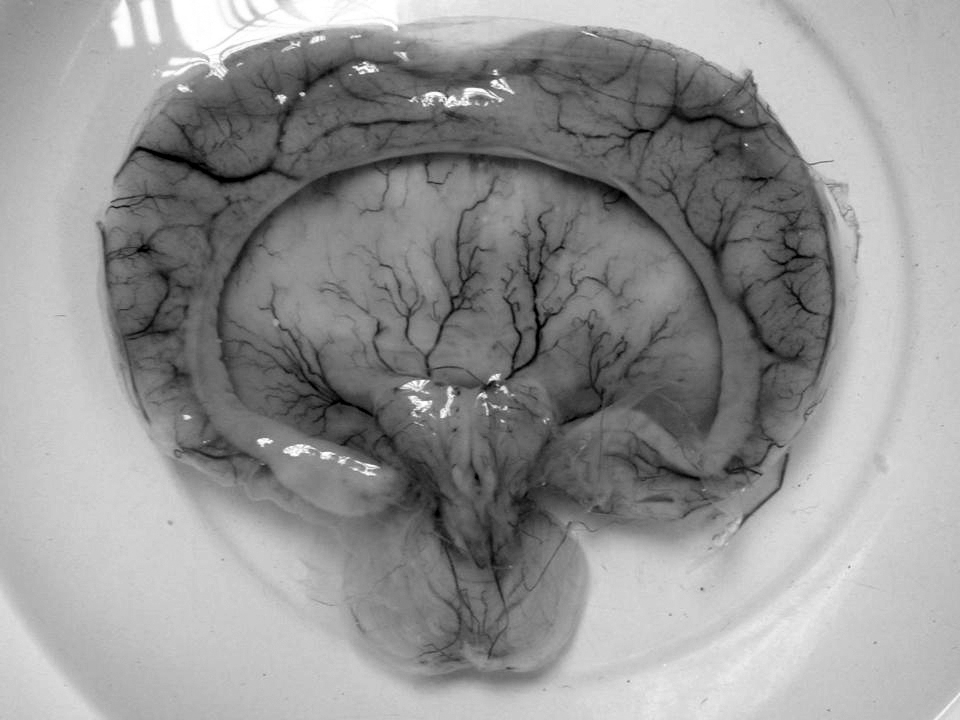
Holoprosencephaly-postaxial polydactyly syndrome associates, in chromosomally normal neonates, holoprosencephaly, severe facial dysmorphism, postaxial polydactyly and other congenital abnormalities, suggestive of trisomy 13 (see this term) Young–Madders syndrome Gross pathology specimen from a case of alobar holoprosencephaly, a clinical manifestation of Young–Madders syndrome first described as a new condition by doctors Young and Madders in 1987. ... All suffered with the features of Young–Madders syndrome, with varying cardiac problems and facial deformities. ... Hydrocephalus and holoprosencephaly were present in all. [4] The publication noted the work of Young and Madders and suggested that the cases were linked, and also identified two cases from a year previously - 1986 - which had until then been diagnosed as Smith–Lemli–Opitz syndrome . The doctors discounted several other similar genetic conditions including Varadi–Papp syndrome and Grote syndrome , and discarded the term 'pseudotrisomy 13 syndrome' as misleading, preferring 'holoprosencephaly-polydactyly syndrome'. [4] Further research [ edit ] After the publications by Verloes, A., S. ... D.; Madders, D. J. (1987). "Unknown syndrome: holoprosencephaly, congenital heart defects, and polydactyly" . ... "Holoprosencephaly–polydactyly ('pseudotrisomy 13') syndrome: a syndrome with features of hydrolethalus and Smith–Lemli–Opitz syndromes.
They concluded that the cases reported by Bachman et al. (1990) were instances of this syndrome and not of the hydrolethalus syndrome (see 236680). The other cases from the literature cited by Bachman et al. (1990) as holoprosencephaly in hydrolethalus syndrome were thought by Cohen and Gorlin (1991) to be in error. Verloes et al. (1991) observed 5 unrelated children with the syndrome and suggested the designation holoprosencephaly-polydactyly syndrome. They commented on the phenotypic overlap not only with the hydrolethalus syndrome but also with lethal acrodysgenital dwarfism (see Smith-Lemli-Opitz syndrome, 270400). ... They reviewed reports of 22 cases of pseudotrisomy 13 syndrome and found postaxial polydactyly in 19 and preaxial polydactyly in 1.
Holoprosencephaly-postaxial polydactyly syndrome associates, in chromosomally normal neonates, holoprosencephaly, severe facial dysmorphism, postaxial polydactyly and other congenital abnormalities, suggestive of trisomy 13 (see this term).
Overview Stevens-Johnson syndrome (SJS) is a rare, serious disorder of the skin and mucous membranes. ... People with cancer, particularly blood cancer, are at increased risk of Stevens-Johnson syndrome. A history of Stevens-Johnson syndrome. ... A family history of Stevens-Johnson syndrome. If an immediate blood relative has had Stevens-Johnson syndrome, you may be at increased risk of getting it too. ... Complications Stevens-Johnson syndrome complications include: Dehydration. ... Preparing for your appointment Stevens-Johnson syndrome is an emergency medical condition.
A number sign (#) is used with this entry because susceptibility to Stevens-Johnson syndrome, allopurinol-induced severe cutaneous adverse reaction, and carbamazepine-induced hypersensitivity syndrome have been associated with HLA-class I alleles (see HLA-A, 142800 and HLA-B, 142830). ... When there is very extensive skin detachment and poor prognosis (death rates of 30 to 40%) the condition is usually called toxic epidermal necrolysis; milder forms are known as Stevens-Johnson syndrome or overlapping Stevens-Johnson syndrome and toxic epidermal necrolysis (Bastuji-Garin et al., 1993). The carbamazepine-induced hypersensitivity syndrome includes symptoms such as rash, fever, eosinophilia, hepatitis, and nephritis. ... In a test for carbamazepine-induced Stevens-Johnson syndrome, the HLA-B*1502 allele should therefore have 100% sensitivity and 97% specificity. ... Carbamazepine is the drug most commonly associated with the syndrome in Asians, accounting for 25 to 33% of cases, whereas only 5 to 6% of Caucasian Stevens-Johnson syndrome cases are caused by it.
A limited form of Stevens-Johnson syndrome/toxic epidermal necrolysis spectrum characterized by destruction and detachment of the skin epithelium and mucous membranes involving less than 10% of the body surface area.
Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) is a very severe reaction, most commonly triggered by medications, that causes skin tissue to die (necrosis) and detach. ... The most common causes of death include sepsis , acute respiratory distress syndrome , and multiple organ failure. Those that survive may experience recurrence (particularly if re-exposed to a trigger) and/or long-term complications involving the skin and affected mucous membranes.
Mapping By linkage analysis in a large Pakistani family with an X-linked mental retardation syndrome, Ahmad et al. (1999) mapped the disorder to the pericentromeric area with a maximum 2-point lod score of 3.86 at zero recombination with marker DXS1106. Reduced recombination events around the centromere prevented precise mapping of the gene. They commented on the X-linked syndromic mental retardation that is associated with gynecomastia and obesity (Wilson-Turner syndrome, or MRXS6; 309585). The MRXS7 candidate region overlaps that of the Wilson-Turner syndrome; however, gynecomastia was absent in the patients of Ahmad et al. (1999). Lubs et al. (1999) used the symbol MRXS7 for the syndromic form of X-linked mental retardation mapped by FISH to Xp11.3-q22 by Ahmad et al. (1997).
A rare, X-linked syndromic intellectual disability disorder characterized by mild to moderate intellectual disability, obesity, hypogonadism, tapering fingers and microphallus with small or undescended testes, localized to Xp11.3-Xq23.
MECP2 duplication syndrome is a severe neurological and developmental disorder. ... Some people with MECP2 duplication syndrome may have autistic features, gastrointestinal problems, and/or mildly distinctive facial features. The syndrome is caused by having an extra copy (duplication) of the MECP2 gene, and inheritance is X-linked. The syndrome almost always occurs in males (who have one X chromosome), but some females with the duplication on one of their two X chromosomes have some signs or symptoms. Rarely, females may have severe signs and symptoms, similar to those in males with the syndrome. Treatment is individualized and based on the signs and symptoms in each person.
The diagnosis of MECP2 duplication syndrome is established in an individual by identification of a heterozygous whole-gene duplication of MECP2 on molecular genetic testing. ... Genetic counseling. MECP2 duplication syndrome is inherited in an X-linked manner. ... Sanlaville et al [2005] Clinical Characteristics Clinical Description MECP2 duplication syndrome is an X-linked disorder, mainly affecting males. ... See OMIM Phenotypic Series: Autosomal dominant ID; Autosomal recessive ID; Nonsyndromic X-linked ID; and Syndromic X-linked ID. Int22h1/int22h2-mediated Xq28 duplication syndrome . ... Cognitive impairment and recurrent infections are common in both syndromes. However, the cognitive impairment in int22h1/int22h2-mediated Xq28 duplication syndrome is less severe and infantile hypotonia, spasticity, and seizures have not been observed.
The same gene is mutated in Rett syndrome (RTT; 312750). X-linked mental retardation with spasticity (300055) is an allelic disorder. ... Meins et al. (2005) reported a boy with psychomotor retardation from birth. He showed features of Rett syndrome, including stereotypic hand movements at age 4 years, loss of purposeful hand movements at 6 years, and autistic features. ... Ramocki et al. (2009) reported 9 boys with MECP2 duplication syndrome from 8 families. All had severe to profound mental retardation, expressive language defects, and autism, with gaze avoidance and avoidance of social interactions. ... Belligni et al. (2010) suggested that MECP2 be evaluated in patients with features of the congenital hypoventilation syndrome (209880). Molecular Genetics In a boy with mental retardation and features of Rett syndrome, Meins et al. (2005) found a submicroscopic duplication of Xq28, including the MECP2 gene (300005.0030). ... Population Genetics The MECP2 duplication syndrome may explain about 1% of cases of X-linked mental retardation, but this number may increase up to 15% when males with specific features, such as progressive spasticity, are studied (Ramocki et al., 2010).
MECP2 duplication syndrome Other names X-linked intellectual disability-hypotonia-recurrent Infections syndrome This condition is due to MECP2 overexpression Specialty Medical genetics MECP2 duplication syndrome ( M2DS ) is a rare disease that is characterized by severe intellectual disability and impaired motor function. ... Jane; Peters, Sarika U. (2010). "TheMECP2duplication syndrome" . American Journal of Medical Genetics Part A . 152A (5): 1079–1088. doi : 10.1002/ajmg.a.33184 . ... PMID 20425814 . ^ a b c d e f g "MECP2 Duplication Syndrome - NORD (National Organization for Rare Disorders)" . ^ Reference, Genetics Home. "MECP2 duplication syndrome" . Genetics Home Reference . ^ "Van Wright Foundation" . Van Wright Foundation . ^ a b Van Esch, H. (2011). "MECP2 Duplication Syndrome" . Molecular Syndromology . 2 (3–5): 128–136. doi : 10.1159/000329580 .
MECP2 duplication syndrome is a condition that occurs almost exclusively in males and is characterized by moderate to severe intellectual disability. ... Individuals with MECP2 duplication syndrome have delayed development of motor skills such as sitting and walking. ... Many individuals with MECP2 duplication syndrome have recurrent respiratory tract infections. ... Frequency The prevalence of MECP2 duplication syndrome is unknown; more than 200 affected individuals have been described in the scientific literature. ... These neuronal changes disrupt normal brain activity, causing the signs and symptoms of MECP2 duplication syndrome. Learn more about the gene associated with MECP2 duplication syndrome MECP2 Inheritance Pattern MECP2 duplication syndrome is inherited in an X-linked pattern.
Vandewalle et al. (2009) noted that the duplicated region of chromosome Xq28 harbored by affected individuals did not contain the MECP2 gene (300005) and is thus distinct from the duplicated region associated with MECP2 duplication syndrome (300260). Clinical features also differ. ... They differed with the conclusion of Vandewalle et al. (2009) that increased GDI1 expression is likely to be responsible for the mental retardation seen in this duplication syndrome, and considered the IKBKG gene (300248), also in the interval, to be likely to play a role in the mental retardation associated with this duplication.
Differential diagnosis Differential diagnoses include Prader-Willi syndrome and Alpha thalassaemia-mental retardation, X linked (ATR-X) syndrome (see these terms). ... Prognosis Malformations do not contribute significantly to the morbidity associated with this syndrome, but early death (before 25 years of age) has been reported in 55% of patients with a MECP2 duplication and a few cases of death during infancy due to infection have also been described.
Mitochondrial DNA-associated Leigh syndrome is a progressive brain disorder that usually appears in infancy or early childhood. ... Mitochondrial DNA-associated Leigh syndrome is a subtype of Leigh syndrome and is caused by changes in mitochondrial DNA . Mutations in at least 11 mitochondrial genes have been found to cause mtDNA-associated Leigh syndrome. This condition has an inheritance pattern known as maternal or mitochondrial inheritance. Because mitochondria can be passed from one generation to the next only through egg cells (not through sperm cells), only females pass mitochondrial DNA-associated Leigh syndrome to their children.
Van Maldergem et al. (2002) suggested that CoQ10 deficiency (607426) can present as Leigh syndrome. Leigh Syndrome Due to Complex IV Deficiency Willems et al. (1977) described deficiency of complex IV, cytochrome c oxidase, in muscle of a child who died at age 6 years of Leigh syndrome. ... The child had the de Toni-Fanconi-Debre renal syndrome and acute neurologic deterioration resembling Leigh syndrome. ... Leigh Syndrome Due to Complex I Deficiency Morris et al. (1996) described complex I deficiency (252010) as an important cause of Leigh syndrome. ... For discussion of a possible association between Leigh syndrome and variation in the IARS2 gene, see 612801.0002. ... It was originally suggested that the biochemical defect in Leigh syndrome was a block in thiamine metabolism.
Maternally inherited Leigh syndrome is a rare subtype of Leigh syndrome (see this term) characterized clinically by encephalopathy, lactic acidosis, seizures, cardiomyopathy, respiratory disorders and developmental delay, with onset in infancy or early childhood, and resulting from maternally-inherited mutations in mitochondrial DNA.
Syndromic recessive X-linked ichthyosis (RXLI) refers to the cases of RXLI (see this term) that are associated with extracutaneous manifestations as part of a syndrome. Epidemiology The prevalence of syndromic RXLI is estimated at 1/50,000-1/150,000. ... Manifestations due to contiguous gene syndrome include neurological abnormalities such as epilepsy and hyposmia, intellectual deficit and/or short stature. This mechanism can be observed in Kallman syndrome, hypergonadotropic hypogonadism, ocular albinism type 1 (see these terms), or hypertrophic pyloric stenosis.
Cauda equina syndrome (CES) refers to a group of symptoms that occur when nerves in the cauda equina (a collection of nerve roots that spread out from the bottom of the spinal cord) become compressed or damaged.
Small intestinal mucosal xanthoma was reported in an individual with CHILD syndrome [Ryan et al 2013]. CK Syndrome CK syndrome (Figure 2) is an X-linked intellectual disability syndrome that affects males. ... Serum concentrations of methyl-sterol and cholesterol are almost always normal in individuals with CHILD syndrome and CK syndrome. Heterozygous females. ... CK syndrome represents the initials of the original proband. CK syndrome was first described by du Souich et al [2009]. ... Differential Diagnosis See Table 2 (CHILD syndrome) and Table 3 (CK syndrome). Table 2.
Mutations in the XPD(ERCC2) gene cause a variety of syndromes; XP, trichothiodystrophy (TTD), or a combination of XP and Cockayne syndrome (XPCS). [19] [20] Both trichothiodystrophy and Cockayne syndrome display features of premature aging, suggesting an association between deficient DNA repair and premature aging (see DNA damage theory of aging ). ... Mutations in the XPG ( ERCC5 ) gene can lead to XP alone, or in combination with Cockayne syndrome (CS), or in combination with infantile lethal cerebro-oculo-facio-skeletal syndrome. [23] Diagnosis [ edit ] Types [ edit ] There are seven complementation groups, plus one variant form: Type Diseases Database OMIM Gene Locus Also known as / description Type A, I, XPA 29877 278700 XPA 9q22.3 Xeroderma pigmentosum group A - the classical form of XP Type B, II, XPB 29878 133510 XPB 2q21 Xeroderma pigmentosum group B Type C, III, XPC 29879 278720 XPC 3p25 Xeroderma pigmentosum group C Type D, IV, XPD 29880 278730 278800 XPD ERCC6 19q13.2-q13.3, 10q11 Xeroderma pigmentosum group D or De Sanctis-Cacchione syndrome (can be considered a subtype of XPD) Type E, V, XPE 29881 278740 DDB2 11p12-p11 Xeroderma pigmentosum group E Type F, VI, XPF 29882 278760 ERCC4 16p13.3-p13.13 Xeroderma pigmentosum group F Type G, VII, XPG 29883 278780 133530 RAD2 ERCC5 13q33 Xeroderma pigmentosum group G and COFS syndrome type 3 Type V, XPV 278750 POLH 6p21.1-p12 Xeroderma pigmentosum variant - these patients have mutation in a gene that codes for a specialized DNA polymerase called polymerase-η (eta) . ... Research directions [ edit ] Research into XP has had two main results: better understanding the disease itself, and also better understanding the normal biological mechanisms involved in DNA repair. [31] Research into XP has produced insights that have been translated into treatments and prevention for cancer. [31] See also [ edit ] DeSanctis–Cacchione syndrome Genetic disorder Biogerontology Cockayne syndrome List of cutaneous conditions List of cutaneous conditions associated with increased risk of nonmelanoma skin cancer Photophobia Senescence References [ edit ] ^ a b c d e f g h i j k l m "Xeroderma pigmentosum" . ... "Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome". Human Mutation . 27 (11): 1092–103. doi : 10.1002/humu.20392 . ... GeneReviews/NCBI/NIH/UW entry on xeroderma pigmentosum Classification D ICD - 10 : Q82.1 ICD - 9-CM : 757.33 OMIM : 278700 MeSH : D014983 DiseasesDB : 14198 External resources MedlinePlus : 001467 eMedicine : derm/462 neuro/399 Patient UK : Xeroderma pigmentosum GeneReviews : Xeroderma Pigmentosum Orphanet : 910 v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation Birthmark v t e Metabolic disease : DNA replication and DNA repair-deficiency disorder DNA replication Separation/initiation: RNASEH2A Aicardi–Goutières syndrome 4 Termination/ telomerase : DKC1 Dyskeratosis congenita DNA repair Nucleotide excision repair Cockayne syndrome / DeSanctis–Cacchione syndrome Thymine dimer Xeroderma pigmentosum IBIDS syndrome MSI / DNA mismatch repair Hereditary nonpolyposis colorectal cancer Muir–Torre syndrome Mismatch repair cancer syndrome MRN complex Ataxia telangiectasia Nijmegen breakage syndrome Other RecQ helicase Bloom syndrome Werner syndrome Rothmund–Thomson syndrome / Rapadilino syndrome Fanconi anemia Li-Fraumeni syndrome Severe combined immunodeficiency v t e Progeroid syndromes DNA repair RecQ-associated Werner syndrome Bloom syndrome Rothmund–Thomson syndrome NER protein-associated Cockayne syndrome Xeroderma pigmentosum Trichothiodystrophy Lamin A/C Hutchinson–Gilford progeria syndrome Restrictive dermopathy Other/related disorders Li–Fraumeni syndrome Rapadilino syndrome Baller–Gerold syndrome DeSanctis–Cacchione syndrome Nijmegen breakage syndrome Fanconi anemia Dyskeratosis congenita Ataxia telangiectasia De Barsy syndrome PIBI(D)S syndrome BIDS syndrome Marfanoid–progeroid–lipodystrophy syndrome See also: DNA replication and repair-deficiency disorder
Phenotype Correlations by Gene For the overall clinical disorders (XP, Cockayne syndrome [CS], trichothiodystrophy [TTD], cerebrooculofacioskeletal [COFS] syndrome, and others [see Figure 1]), the clinical phenotypes are related within broad groups to the specific gene that is mutated (see Figure 1 and Table 2). ... One individual with a homozygous ERCC1 pathogenic variant had severe Cockayne syndrome type II and died at age 2.5 years [Kashiyama et al 2013]. 3. ... Differential Diagnosis Xeroderma pigmentosum (XP), XP with neurologic abnormalities, Cockayne syndrome (CS), the XP/CS complex, trichothiodystrophy (TTD), the XP/TTD complex, cerebrooculofacioskeletal (COFS) syndrome, COFS/TTD, CS/TTD complex, and the UV-sensitive syndrome [Horibata et al 2004, Berneburg & Kraemer 2007, Kraemer et al 2007, Stefanini & Kraemer 2008, Kraemer & Ruenger 2012, Ruenger et al 2012] represent ten genetic diseases that exhibit cutaneous photosensitivity caused by defective nucleotide excision repair (NER). ... Meira et al [2000], Graham et al [2001] 4. Two individuals with Cockayne syndrome and one individual with phenotypic features of Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia had biallelic pathogenic variants in either ERCC1 or ERCC4 [Kashiyama et al 2013]. 5. ... These other diseases include the following: Rothmund-Thomson syndrome and the allelic disorder Baller-Gerold syndrome Hartnup disorder (OMIM 234500), a disorder of amino acid absorption resulting from biallelic pathogenic variants in SLC6A19 , a non-polar amino acid transporter.
History Moshell et al. (1983) defined complementation group H xeroderma pigmentosum on the basis of a single patient who had both xeroderma pigmentosum and Cockayne syndrome. Johnson et al. (1989) found that hybrids between XPD cells and cells from groups A, B, C, E, F, G, and I showed cross-correction. ... INHERITANCE - Autosomal recessive HEAD & NECK Head - Microcephaly Ears - Sensorineural deafness Eyes - Photophobia - Conjunctivitis - Keratitis - Ectropion - Entropion - Microphthalmia (uncommon) - Infantile cataract - Decreased best corrected visual acuity - Dry eye syndrome (in some patients) - Ocular surface fluorescein staining (in some patients) - Corneal neovascularization (in some patients) SKIN, NAILS, & HAIR Skin - Skin photosensitivity - Early onset skin cancer (basal cell, squamous cell and malignant melanoma) - Early freckle-like lesions in exposed areas - Poikiloderma - Increased/decreased skin pigment - Skin atrophy - Telangiectasia - Actinic keratoses - Angiomas - Keratoacanthomas NEUROLOGIC Central Nervous System - Mental deterioration - Low intelligence - Hyporeflexia - Spasticity - Ataxia - Choreoathetosis NEOPLASIA - Early onset skin cancer (basal cell, squamous cell and malignant melanoma) LABORATORY ABNORMALITIES - Defective DNA repair after ultraviolet radiation damage MISCELLANEOUS - Later onset of neurologic features MOLECULAR BASIS - Caused by mutation in the excision repair cross complementing rodent repair deficiency, complementation group-2 gene (ERCC2, 126340.0001 ) ▲ Close
Some patients develop neurologic symptoms or a more severe clinical phenotype known as de Sanctis-Cacchione syndrome (278800) (Satokata et al., 1992). ... For each of the 7 types, they tabulated the occurrence of clinical manifestations in 4 areas, skin cancer, neurologic abnormality, symptoms of Cockayne syndrome (see CSA, 216400), and trichothiodystrophy (601675), as well as the levels of unscheduled DNA synthesis after UV exposure and of UV sensitivity. ... Some aspect of DNA repair mechanisms is deficient in 4 other inherited diseases: ataxia-telangiectasia (AT; 208900), Fanconi anemia, Bloom syndrome (BLM; 210900), and Cockayne syndrome. ... They believed that xeroderma pigmentosum with neurologic abnormalities (de Sanctis-Cacchione syndrome) was first reported in 1883 by Albert Neisser of Breslau, Germany (who also discovered the bacterial cause of gonorrhea, the agent named Neisseria).
Clinical Features In addition to the usual severe, recessively inherited xeroderma pigmentosum (278700-278780), the existence of a milder form behaving as a dominant was claimed by Anderson and Begg (1950) who described 11 affected persons in 5 sibships of 4 generations of a Scottish family by the name of MacPherson. The patients showed freckling and multiple skin cancers as in the recessive form but did not get into trouble as early in life and survived longer. Indeed, Anderson and Begg examined 1 affected member of the family who was 74 years of age. No affected member was said to have died of the disease. Sedano (1984) raised doubts about this family. In the Scottish family, Anderson personally examined 7 members: 'Mr. George MacPherson, sen., his wife, George, jun., Harold, Lena, Douglas, and the son of George, jun.
These include acquired microcephaly, diminished or absent deep tendon stretch reflexes, progressive sensorineural hearing loss, spasticity, ataxia, seizures and progressive cognitive impairment. De Sanctis-Cacchione syndrome is a term that was originally attributed to XP cases with severe neurological abnormalities but it is no longer in general use. ... Differential diagnosis Differential diagnoses include trichothiodystrophy, Cockayne syndrome, cerebrooculofacioskeletal syndrome (COFS), UV-sensitive syndrome, erythropoietic protoporphyria, and Rothmund-Thomson syndrome (see these terms).
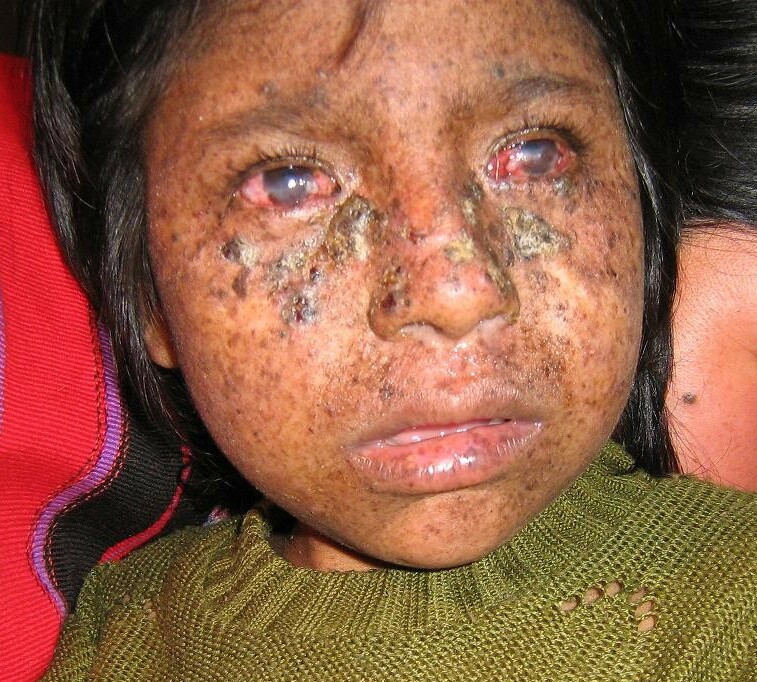
Xeroderma pigmentosum (XP) causes the skin and eyes to be extra sensitive to exposure to ultraviolet radiation from the sun and other sources. Symptoms begin in early childhood. People with XP can develop bad sunburns, blistering, and freckling in response to sunlight. The eyes may develop light sensitivity, corneal clouding, and swelling. Some people with XP have nervous system involvement as well. People with XP are at very high risk of developing skin cancer and other types of cancers. XP is caused by variants in one of at least nine genes involved in repairing damaged DNA.
Pathogenesis XP patients are susceptible to UV-induced freckling and malignant skin changes, whereas Cockayne syndrome (see, e.g., 216400) patients do not have increased susceptibility to these lesions. Seguin et al. (1988) studied the frequency of ultraviolet light-induced chromosomal breaks in lymphoblastoid cell lines from patients with Cockayne syndrome and from patients with xeroderma pigmentosum.
Description For a general discussion of xeroderma pigmentosum, see XPA (278700), and of Cockayne syndrome, see CSA (216400). Cleaver (1990) provided a review of the causes of xeroderma pigmentosum. ... Scott et al. (1993) described 2 brothers with XP/Cockayne syndrome complex who fell into the complementation group B category. ... Molecular Genetics In a patient with XPB/Cockayne syndrome reported by Robbins et al. (1974), Weeda et al. (1990) identified a heterozygous mutation in the ERCC3 gene (133510.0001). ... In 2 brothers with XPB/Cockayne syndrome complex reported by Scott et al. (1993), Vermeulen et al. (1994) identified a heterozygous mutation in the ERCC3 gene (133510.0002). ... The patient had combined clinical signs of XP and Cockayne syndrome, which Hwang et al. (1996) concluded were consistent with a combined defect in DNA repair and transcription.
Some patients with XPF may develop neurologic impairment or growth defects, and are then classified as having Cockayne syndrome (summary by Kashiyama et al., 2013). ... Single skin cancers occurred in only 3 of the 11 patients, with an average age of 52 years for their first skin malignancy. Cockayne Syndrome Moriwaki et al. (1993) reported a 48-year-old Japanese man with xeroderma pigmentosum associated with mental retardation, cerebral atrophy, and cerebellar ataxia. ... Kashiyama et al. (2013) reported a 16-year-old boy (CS1USAU) with Cockayne syndrome. He developed normally for the first year without obvious abnormalities, except for the microcephaly. ... A second unrelated patient (XPCS1CD) had a more complex phenotype comprising XPF, Cockayne syndrome, and Fanconi anemia (see FANCQ; 615272). ... In 2 unrelated patients with XPF and Cockayne syndrome, Kashiyama et al. (2013) identified compound heterozygous mutations in the ERCC4 gene (133520.0008-133520.0010).
A number sign (#) is used with this entry because xeroderma pigmentosum complementation group E is caused by homozygous mutation in the DDB2 gene (600811) on chromosome 11p11. For background information on xeroderma pigmentosum, see 278700. Clinical Features Kondo et al. (1988) studied 3 Japanese patients, aged 50, 42, and 41 years, with mild XP symptoms and no neurologic abnormalities. Two had developed basal cell carcinomas at ages 46 and 41 years. Chu and Chang (1988) studied cells from 2 second-cousin patients with group E xeroderma pigmentosum who had a skin disease less severe than that seen in patients of several other complementation groups (de Weerd-Kastelein et al., 1974). Using an extension of the gel electrophoresis binding assay, Chu and Chang (1988) identified at least 1 nuclear factor that binds to DNA damaged by ultraviolet radiation or the antitumor drug cisplatin, but is notably absent in group E cells. The defect was not the result of a failure of protein transport to the nucleus since binding activity was absent in both the nuclear and cytoplasmic extracts.