A number sign (#) is used with this entry because of evidence that Jaberi-Elahi syndrome (JABELS) is caused by homozygous mutation in the GTPBP2 gene (607434) on chromosome 6p21.
Contents 1 Signs and symptoms 2 Cause 3 Diagnosis 3.1 Types 3.2 Primary 3.3 Secondary 4 Treatment 5 See also 6 Other animals 7 References 8 External links Signs and symptoms [ edit ] It can be asymptomatic , but these symptoms may be present: [ citation needed ] Fatigue Headache High blood pressure Hypokalemia Hypernatraemia Hypomagnesemia Intermittent or temporary paralysis Muscle spasms Muscle weakness Numbness Polyuria Polydipsia Tingling Metabolic alkalosis [2] Cause [ edit ] The causes of primary hyperaldosteronism are adrenal hyperplasia and adrenal adenoma (Conn's syndrome). These cause hyperplasia of aldosterone-producing cells of the adrenal cortex resulting in primary hyperaldosteronism. ... Primary [ edit ] Main article: Primary aldosteronism Primary aldosteronism (hyporeninemic hyperaldosteronism) was previously thought to be most commonly caused by an adrenal adenoma , termed Conn's syndrome . However, recent studies have shown that bilateral idiopathic adrenal hyperplasia is the cause in up to 70% of cases. ... Other causes can come from the tubules: low reabsorption of sodium (as seen in Bartter and Gitelman syndromes) will lead to hypovolemia/hypotension, which will activate the renin–angiotensin system (RAAS). [ citation needed ] Secondary hyperaldosteronism can also be caused by excessive ingestion of licorice or other members of the Glycyrrhiza genus of plants that contain the triterpenoid saponin glycoside known as glycyrrhizin . ... PMID 11073536 . ^ Sabbadin C, Armanini D (September 2016). "Syndromes that mimic an excess of mineralocorticoids".
A rare idiopathic interstitial pneumonia characterized by extensive, diffuse intra-alveolar accumulation of pigment-laden macrophages, most commonly associated with long-term exposure to tobacco smoke. Patients present with slowly progressive shortness of breath on exertion and chronic cough with bilateral crackles. Digital clubbing is also frequently observed. Pulmonary function test reveals a restrictive pattern. Computed tomography typically shows diffuse ground-glass opacities with subpleural and lower zone predominance.
Description Interstitial lung disease (ILD), or pneumonitis, is a heterogeneous group of disorders characterized pathologically by expansion of the interstitial compartment of the lung by inflammatory cells. Fibrosis occurs in many cases (Visscher and Myers, 2006). Desquamative interstitial pneumonitis (DIP) was originally described as a pathologic entity by Liebow et al. (1965). Lung biopsy shows diffuse and uniform filling of alveoli by clusters of cells which Liebow et al. (1965) speculated to be 'desquamated pneumocytes.' Since then, these cells have been shown primarily to be pigmented alveolar macrophages. Other features include thickened alveolar septa with an infiltrate of inflammatory cells and plump, cuboidal type II pneumocytes.
Symptoms may come and go depending on whether the person receives treatment, and whether the treatment takes effect. [ citation needed ] Causes [ edit ] Thyroid autoimmunity is familial. [1] The disease is said to be inherited as a dominant trait since it has been reported that as many as fifty percent of the first degree relatives of patients with some type of autoimmune thyroiditis present with thyroid antibodies in serum. [1] Some studies have even related it to chromosome 21 because of its high correlation with patients with Down syndrome and familial Alzheimer disease. This theory is controversial, since patients with Turner syndrome also present a high prevalence of autoimmune thyroiditis (up to fifty percent). [1] High iodine consumption [ edit ] Autoimmune thyroiditis has a higher prevalence in societies that have a higher intake of iodine in their diet, such as the United States and Japan. ... Retrieved 3 December 2020 . ^ https://www.btf-thyroid.org/thyroiditis External links [ edit ] Classification D ICD - 10 : E06.3 MeSH : D013967 SNOMED CT : 66944004 v t e Thyroid disease Hypothyroidism Iodine deficiency Cretinism Congenital hypothyroidism Myxedema Myxedema coma Euthyroid sick syndrome Signs and symptoms Queen Anne's sign Woltman sign Thyroid dyshormonogenesis Pickardt syndrome Hyperthyroidism Hyperthyroxinemia Thyroid hormone resistance Familial dysalbuminemic hyperthyroxinemia Hashitoxicosis Thyrotoxicosis factitia Thyroid storm Graves' disease Signs and symptoms Abadie's sign of exophthalmic goiter Boston's sign Dalrymple's sign Stellwag's sign lid lag Griffith's sign Möbius sign Pretibial myxedema Graves' ophthalmopathy Thyroiditis Acute infectious Subacute De Quervain's Subacute lymphocytic Palpation Autoimmune /chronic Hashimoto's Postpartum Riedel's Enlargement Goitre Endemic goitre Toxic nodular goitre Toxic multinodular goiter Thyroid nodule Colloid nodule
A rare idiopathic interstitial pneumonia characterized by temporally uniform alveolar and interstitial mononuclear cell inflammation (cellular type) and/or fibrosis of the alveolar walls (fibrotic type) with preserved alveolar architecture. Other types of interstitial lung disease must be excluded. Symptoms are non-specific and include dyspnea, cough, and often constitutional symptoms such as fever and fatigue. Pulmonary function test reveals a restrictive pattern. Computed tomography shows predominantly lower lobe subpleural reticular changes, traction bronchiectasis, and ground-glass opacities. The cellular type of the disease is less common but carries a better prognosis.
A number sign (#) is used with this entry because argininosuccinic aciduria is caused by homozygous mutation in the gene encoding argininosuccinate lyase (ASL; 608310) on chromosome 7q11. Description Argininosuccinic aciduria is an autosomal recessive disorder of the urea cycle. Urea cycle disorders are characterized by the triad of hyperammonemia, encephalopathy, and respiratory alkalosis. Five disorders involving different defects in the biosynthesis of the enzymes of the urea cycle have been described: ornithine transcarbamylase deficiency (311250), carbamyl phosphate synthetase deficiency (237300), argininosuccinate synthetase deficiency, or citrullinemia (215700), argininosuccinate lyase deficiency, and arginase deficiency (207800). Erez (2013) reviewed argininosuccinic aciduria and progress in understanding it as a monogenic disorder that, like other inborn errors of metabolism, manifests as a multifactorial disorder at the phenotypic level.
Summary Clinical characteristics. Deficiency of argininosuccinate lyase (ASL), the enzyme that cleaves argininosuccinic acid to produce arginine and fumarate in the fourth step of the urea cycle, may present as a severe neonatal-onset form and a late-onset form: The severe neonatal-onset form is characterized by hyperammonemia within the first few days after birth that can manifest as increasing lethargy, somnolence, refusal to feed, vomiting, tachypnea, and respiratory alkalosis. Absence of treatment leads to worsening lethargy, seizures, coma, and even death. In contrast, the manifestations of late-onset form range from episodic hyperammonemia triggered by acute infection or stress to cognitive impairment, behavioral abnormalities, and/or learning disabilities in the absence of any documented episodes of hyperammonemia. Manifestations of ASL deficiency (ASLD) that appear to be unrelated to the severity or duration of hyperammonemic episodes: Neurocognitive deficiencies (attention-deficit/hyperactivity disorder, developmental delay, seizures, and learning disability) Liver disease (hepatitis, cirrhosis) Trichorrhexis nodosa (coarse brittle hair that breaks easily) Systemic hypertension Diagnosis/testing. Elevated plasma ammonia concentration (>100 µmol/L), elevated plasma citrulline concentration (usually 100-300 µmol/L), and elevated argininosuccinic acid in the plasma or urine establish the diagnosis of ASLD.
Argininosuccinic aciduria is an inherited disorder that causes ammonia to accumulate in the blood. Ammonia, which is formed when proteins are broken down in the body, is toxic if the levels become too high. The nervous system is especially sensitive to the effects of excess ammonia. Argininosuccinic aciduria usually becomes evident in the first few days of life. An infant with argininosuccinic aciduria may be lacking in energy (lethargic) or unwilling to eat, and have poorly controlled breathing rate or body temperature.
A rare, genetic disorder of urea cycle metabolism typically characterized by either a severe, neonatal-onset form that manifests with hyperammonemia accompanied with vomiting, hypothermia, lethargy and poor feeding in the first few days of life, or late-onset forms that manifest with stress- or infection-induced episodic hyperammonemia or, in some, behavioral abnormalities and/or learning disabilities, or chronic liver disease. Patients often manifest liver dysfunction. Epidemiology The prevalence at birth of argininosuccinic aciduria (ASA) ranges between 1/70,000-218,000 worldwide. Clinical description ASA can have a variable clinical picture with either a neonatal-onset or a late-onset (at any age outside the newborn period). Neonates with severe neonatal-onset ASA usually appear normal during the first 24-48 hours after birth but within a few days present with severe hyperammonemia manifesting with lethargy, somnolence, refusal to feed, vomiting, tachypnea and respiratory alkalosis. If untreated, worsening lethargy, seizures, coma and death may occur.
Argininosuccinic aciduria is an inherited disorder that causes ammonia to accumulate in the blood. Ammonia, which is formed when proteins are broken down in the body, is toxic if the levels become too high. The nervous system is especially sensitive to the effects of excess ammonia. Argininosuccinic aciduria usually becomes evident in the first few days of life. An infant with argininosuccinic aciduria may be lacking in energy (lethargic) or unwilling to eat, and have a poorly controlled breathing rate or body temperature.
External links [ edit ] Classification D ICD - 10 : E72.2 ICD - 10-CM : E72.21 OMIM : 207800 MeSH : D020162 DiseasesDB : 29677 External resources eMedicine : ped/132 GeneReviews : Arginase Deficiency Orphanet : 90 Scholia has a topic profile for Argininemia . v t e Inborn error of amino acid metabolism K → acetyl-CoA Lysine /straight chain Glutaric acidemia type 1 type 2 Hyperlysinemia Pipecolic acidemia Saccharopinuria Leucine 3-hydroxy-3-methylglutaryl-CoA lyase deficiency 3-Methylcrotonyl-CoA carboxylase deficiency 3-Methylglutaconic aciduria 1 Isovaleric acidemia Maple syrup urine disease Tryptophan Hypertryptophanemia G G→ pyruvate → citrate Glycine D-Glyceric acidemia Glutathione synthetase deficiency Sarcosinemia Glycine → Creatine : GAMT deficiency Glycine encephalopathy G→ glutamate → α-ketoglutarate Histidine Carnosinemia Histidinemia Urocanic aciduria Proline Hyperprolinemia Prolidase deficiency Glutamate / glutamine SSADHD G→ propionyl-CoA → succinyl-CoA Valine Hypervalinemia Isobutyryl-CoA dehydrogenase deficiency Maple syrup urine disease Isoleucine 2-Methylbutyryl-CoA dehydrogenase deficiency Beta-ketothiolase deficiency Maple syrup urine disease Methionine Cystathioninuria Homocystinuria Hypermethioninemia General BC / OA Methylmalonic acidemia Methylmalonyl-CoA mutase deficiency Propionic acidemia G→ fumarate Phenylalanine / tyrosine Phenylketonuria 6-Pyruvoyltetrahydropterin synthase deficiency Tetrahydrobiopterin deficiency Tyrosinemia Alkaptonuria / Ochronosis Tyrosinemia type I Tyrosinemia type II Tyrosinemia type III / Hawkinsinuria Tyrosine → Melanin Albinism : Ocular albinism ( 1 ) Oculocutaneous albinism ( Hermansky–Pudlak syndrome ) Waardenburg syndrome Tyrosine → Norepinephrine Dopamine beta hydroxylase deficiency reverse: Brunner syndrome G→ oxaloacetate Urea cycle / Hyperammonemia ( arginine aspartate ) Argininemia Argininosuccinic aciduria Carbamoyl phosphate synthetase I deficiency Citrullinemia N-Acetylglutamate synthase deficiency Ornithine transcarbamylase deficiency / translocase deficiency Transport / IE of RTT Solute carrier family : Cystinuria Hartnup disease Iminoglycinuria Lysinuric protein intolerance Fanconi syndrome : Oculocerebrorenal syndrome Cystinosis Other 2-Hydroxyglutaric aciduria Aminoacylase 1 deficiency Ethylmalonic encephalopathy Fumarase deficiency Trimethylaminuria v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
Arginase deficiency is an inherited metabolic disease in which the body is unable to process arginine (a building block of protein). It belongs to a group of disorders known as urea cycle disorders . These occur when the body's process for removing ammonia is disrupted, which can cause ammonia levels in the blood to rise (hyperammonemia). In most cases, symptoms appear between the ages of one and three years. Symptoms may include feeding problems, vomiting, poor growth, seizures, and stiff muscles with increased reflexes (spasticity). People with arginase deficiency may also have developmental delay, loss of developmental milestones, and intellectual disability.
A number sign (#) is used with this entry because argininemia is caused by homozygous or compound heterozygous mutation in the arginase-1 gene (ARG1; 608313) on chromosome 6q23. Description Arginase deficiency is an autosomal recessive inborn error of metabolism caused by a defect in the final step in the urea cycle, the hydrolysis of arginine to urea and ornithine. Urea cycle disorders are characterized by the triad of hyperammonemia, encephalopathy, and respiratory alkalosis. Five disorders involving different defects in the biosynthesis of the enzymes of the urea cycle have been described: ornithine transcarbamylase deficiency (311250), carbamyl phosphate synthetase deficiency (237300), argininosuccinate synthetase deficiency, or citrullinemia (215700), argininosuccinate lyase deficiency (207900), and arginase deficiency. Clinical Features Terheggen et al. (1969, 1970) described 2 sisters, aged 18 months and 5 years, with spastic paraplegia, epileptic seizures, and severe mental retardation.
Arginase deficiency is an inherited disorder that causes the amino acid arginine (a building block of proteins) and ammonia to accumulate gradually in the blood. Ammonia, which is formed when proteins are broken down in the body, is toxic if levels become too high. The nervous system is especially sensitive to the effects of excess ammonia. Arginase deficiency usually becomes evident by about the age of 3. It most often appears as stiffness, especially in the legs, caused by abnormal tensing of the muscles (spasticity). Other symptoms may include slower than normal growth, developmental delay and eventual loss of developmental milestones, intellectual disability, seizures, tremor, and difficulty with balance and coordination (ataxia).
A rare autosomal recessive amino acid metabolism disorder characterized clinically by variable degrees of hyperammonemia, developing from about 3 years of age, and leading to progressive loss of developmental milestones and spasticity in the absence of treatment.
Ho et al [2019] are the first to document the use of sodium phenylbutyrate throughout two sequential pregnancies in a woman with HHH syndrome: In the first pregnancy sodium phenylbutyrate (5.5 g/4x/day) was used as maintenance therapy.
Overview Claudication is pain caused by too little blood flow to muscles during exercise. Most often this pain occurs in the legs after walking at a certain pace and for a certain amount of time — depending on the severity of the condition. The condition is also called intermittent claudication because the pain usually isn't constant. It begins during exercise and ends with rest. As claudication worsens, however, the pain may occur during rest. Claudication is technically a symptom of disease, most often peripheral artery disease, a narrowing of arteries in the limbs that restricts blood flow.
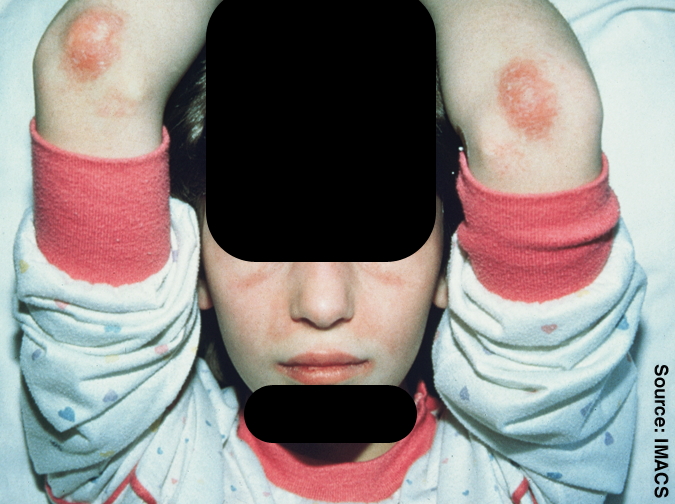
The course of JDM is highly variable: some patients go into remission within 2 to 3 years, others have a cyclic course marked by relapse, while some have ulcerative or chronic disease. Macrophage activation syndrome, a severe sometimes life-threatening condition, has been described in some children diagnosed with JDM.
Juvenile dermatomyositis has some similarities to adult dermatomyositis and polymyositis . It typically affects children ages 2 to 15 years, with symptoms that include weakness of the muscles close to the trunk of the body, inflammation, edema , muscle pain, fatigue, skin rashes, abdominal pain, fever, and contractures. Children with juvenile dermatomyositis may have difficulty swallowing and breathing, and the heart may also be affected. About 20 to 30 percent of children with juvenile dermatomyositis develop calcium deposits in the soft tissue. Affected children may not show higher than normal levels of the muscle enzyme creatine kinase in their blood but have higher than normal levels of other muscle enzymes.
Hypersensitivity drug reactions differ from drug toxicity reactions in that drug toxicity reactions result from the pharmacological action of a drug, are dose-related, and can occur in any treated individual (see nonsteroidal anti-inflammatory drugs section on adverse reactions for NSAID-induced toxic reactions); hypersensitivity reactions are idiosyncratic reactions to a drug. [1] Although the term NSAID was introduced to signal a comparatively low risk of adverse effects, [2] NSAIDs do evoke a broad range of hypersensitivity syndromes. These syndromes have recently been classified by the European Academy of Allergy and Clinical Immunology Task Force on NSAIDs Hypersensitivity. [3] Contents 1 Classification 2 References Classification [ edit ] The classification organizes the hypersensitivity reactions to NSAIDs into the following five categories: NSAIDs-exacerbated respiratory disease (NERD) is an acute (immediate to several hours) exacerbation of bronchoconstriction and other symptoms of asthma (see aspirin-induced asthma ) in individuals with a history of asthma and/or nasal congestion , rhinorrhea or other symptoms of rhinitis and sinusitis in individuals with a history of rhinosinusitis after ingestion of various NSAIDs, particularly those that act by inhibiting the COX-1 enzyme. ... SNIDR are most commonly skin reactions that may be relatively mild moderately severe such as maculopapular rash , fixed drug eruptions , photosensitivity reactions , delayed urticaria , and contact dermatitis or extremely severe such as the DRESS syndrome , acute generalized exanthematous pustulosis , the Stevens–Johnson syndrome , and toxic epidermal necrolysis (also termed Lyell's syndrome).
Complications [ edit ] Intestinal malrotation can lead to a number of disease manifestations and complications such as: [ citation needed ] Acute midgut volvulus Chronic midgut volvulus Acute duodenal obstruction Chronic duodenal obstruction Short bowel syndrome , in cases of volvulus with intestinal necrosis Death, in cases of volvulus with pan-necrosis of the bowel, severe septic shock or hypovolemic shock Malabsorption Chronic motility issues Internal herniation Superior mesenteric artery syndrome Causes [ edit ] Diagram showing the process by which the intestine rotates and herniates during normal development. ... External links [ edit ] Classification D ICD - 10 : Q43.3 ICD - 9-CM : 751.4 OMIM : 193250 MeSH : C562456 C562456, C562456 External resources eMedicine : ped/1200 v t e Congenital malformations and deformations of digestive system Upper GI tract Tongue , mouth and pharynx Cleft lip and palate Van der Woude syndrome tongue Ankyloglossia Macroglossia Hypoglossia Esophagus EA/TEF Esophageal atresia: types A, B, C, and D Tracheoesophageal fistula: types B, C, D and E esophageal rings Esophageal web (upper) Schatzki ring (lower) Stomach Pyloric stenosis Hiatus hernia Lower GI tract Intestines Intestinal atresia Duodenal atresia Meckel's diverticulum Hirschsprung's disease Intestinal malrotation Dolichocolon Enteric duplication cyst Rectum / anal canal Imperforate anus Rectovestibular fistula Persistent cloaca Rectal atresia Accessory Pancreas Annular pancreas Accessory pancreas Johanson–Blizzard syndrome Pancreas divisum Bile duct Choledochal cysts Caroli disease Biliary atresia Liver Alagille syndrome Polycystic liver disease
The propositus, his 2 sons and 3 daughters, and his 2 grandchildren demonstrated this midgut malrotation syndrome. The midgut volvulus caused great discomfort.
Patients most typically present in the neonatal period with midgut volvulus, which can lead to short bowel syndrome or even death. Signs and symptoms include bilious vomiting, feeding intolerance, failure to thrive, constipation, bloody stools, or intermittent apnea.
A rare, autosomal recessive, organic aciduria that is characterized by variable clinical presentation ranging from acute neonatal onset of metabolic decompensation to later onset of chronic, non-specific manifestations including failure to thrive and/or developmental delay. All patients are prone to intermittent, acute metabolic decompensation. During metabolic episodes, urine analysis demonstrates elevated isovaleric acid derivatives. Epidemiology Accurate data on the prevalence is not readily available. Best estimates come from newborn screening studies that estimate prevalence at birth between 1/50,000-150,000.
Isovaleric acidemia (IVA) occurs when the body cannot breakdown certain parts of the proteins found in food. This can cause a build-up of toxic substances which can lead to bouts of serious illness known as metabolic crises. There are two types of IVA. The acute, neonatal type has more severe symptoms that begin in the newborn period. In the chronic, intermittent type symptoms appear during childhood and can come and go. Symptoms include poor feeding, tremor, vomiting, low muscle tone, and lack of energy (lethargy).
A number sign (#) is used with this entry because of evidence that isovaleric acidemia (IVA) is caused by homozygous mutation in the isovaleryl CoA dehydrogenase gene (IVD; 607036) on chromosome 15q15. Description Isovaleric acidemia is an inborn error of leucine metabolism caused by a deficiency of isovaleryl-CoA dehydrogenase. It can present with severe neonatal ketoacidosis leading to death, but in milder cases recurrent episodes of ketoacidosis of varying degree occur later in infancy and childhood (summary by Vockley et al., 1991). Clinical Features Two forms of isovaleric acidemia are recognized: the acute neonatal form, leading to massive metabolic acidosis from the first days of life and rapid death (e.g., Newman et al., 1967), and a chronic form in which periodic attacks of severe ketoacidosis occur with asymptomatic intervening periods (e.g., Tanaka et al., 1966). Budd et al. (1967) observed brother and sister who, before the age of 6 months, showed retarded psychomotor development, a peculiar odor resembling sweaty feet, an aversion to dietary protein, and pernicious vomiting, leading to acidosis and coma.
Isovaleric acidemia is a rare disorder in which the body is unable to properly break down a particular protein building block (amino acid ). The condition is classified as an organic acid disorder, which is a condition that leads to an abnormal buildup of particular acids known as organic acids. Abnormal levels of organic acids in the blood (organic acidemia), urine (organic aciduria), and tissues can be toxic and can cause serious health problems. Normally, the body breaks down proteins from food into smaller parts called amino acids. Amino acids can be further processed to provide energy for growth and development.
If the genes for peroneal muscular atrophy and Friedreich ataxia are closely situated on the X chromosome, deletion is another possible explanation for the finding in this family, namely, a 'contiguous gene syndrome' (Schmickel, 1986). In the kindred reported by Biemond (1928), some individuals had Charcot-Marie-Tooth disease (in a pedigree pattern consistent with X-linked inheritance), whereas 2 females of 1 sibship had Friedreich ataxia. ... Skel - Pes cavus - Scoliosis - Hammer toe Limbs - Foot drop - Steppage gait - Pes cavus Neuro - Neuropathy - Areflexia - Ulnar nerve enlargement - Mild to moderate distal limb sensory loss - Cerebellar ataxia - Dysarthria - Nystagmus - Incoordined limb movements - Diminished or absent tendon reflexes - Babinski sign - Impaired position sense - Impaired vibratory sense - Hypoactive knee and ankle jerks Inheritance - X-linked - ? contiguous gene syndrome Metabolic - Diabetes mellitus - Diabetic ketosis Cardiac - Symmetric, concentric, hypertrophic cardiomyopathy - Congestive heart failure - Muscular subaortic stenosis Muscle - Tibialis weakness and atrophy - Anterior peroneal weakness and atrophy - Intrinsic hand muscles weakness and atrophy Misc - Onset before adolescence Lab - Abnormal motor and sensory nerve conduction - Abnormal spinocerebellar tracts, dorsal columns, pyramidal tracts, cerebellum and medulla - Abnormal EKG - Abnormal echocardiogram - Low pyruvate carboxylase activity in liver and cultured fibroblasts - Mitochondrial malic enzyme reduced ▲ Close
The patients showed some clinical and biochemical similarities with the Wiskott-Aldrich syndrome (301000) but important differences as well. ... The autoradiographs of the lymphocytes in Wiskott-Aldrich syndrome were similar to those in this disorder, but in WAS glycoprotein-115 is abnormal in both platelets and lymphocytes (Remold-O'Donnell et al., 1984).
Partial resistance to progesterone, on the other hand, would be expected to be associated with various degrees of incomplete maturation of the endometrium, perhaps expressed clinically as infertility or early abortions. The syndrome would present with the clinical and histologic picture of a luteal phase defect in which the life span of the corpus luteum and the plasma progesterone concentrations would be normal or elevated. ... Molecular Genetics Although the progesterone resistance syndrome may be due to mutations in the progesterone receptor gene, Chrousos (2002) stated that none such had been identified to that time.
Find sources: "Cutis marmorata" – news · newspapers · books · scholar · JSTOR ( September 2014 ) ( Learn how and when to remove this template message ) Cutis marmorata Cutis marmorata in a patient with Type I decompression sickness (DCS) Specialty Dermatology Cutis marmorata (from Latin marmor , "marble") is a benign skin condition which, if persistent, occurs in Cornelia de Lange syndrome , trisomy 13 and trisomy 18 syndromes. [1] When a newborn infant is exposed to low environmental temperatures, an evanescent , lacy, reticulated red and/or blue cutaneous vascular pattern appears over most of the body surface.