Using the same method, in 1999–2004, myopia prevalence was estimated to have climbed to 42%. [100] A study of 2,523 children in grades 1 to 8 (age, 5–17 years) found nearly one in 10 (9%) have at least −0.75 diopters of myopia. [101] In this study, 13% had at least +1.25 D hyperopia (farsightedness), and 28% had at least 1.00-D difference between the two principal meridians (cycloplegic autorefraction) of astigmatism . ... October 2010. Archived from the original on 28 July 2016 . Retrieved 30 July 2016 . ^ a b c d e f g h i j k l m n o Foster PJ, Jiang Y (February 2014). "Epidemiology of myopia" . Eye . 28 (2): 202–8. doi : 10.1038/eye.2013.280 . ... "Myopia, an underrated global challenge to vision: where the current data takes us on myopia control" . Eye . 28 (2): 142–6. doi : 10.1038/eye.2013.256 . ... Ophthalmic & Physiological Optics . 28 (2): 103–14. doi : 10.1111/j.1475-1313.2008.00550.x .
Rare isolated myopia is a rare, genetic, refraction anomaly disorder characterized by non-syndromic severe myopia, which may be associated with cataract and vitreoretinal degeneration (retinal detachment) that may lead to blindness.
Overview Nearsightedness (myopia) is a common vision condition in which near objects appear clear, but objects farther away look blurry. It occurs when the shape of the eye — or the shape of certain parts of the eye — causes light rays to bend (refract) inaccurately. Light rays that should be focused on nerve tissues at the back of the eye (retina) are focused in front of the retina. Nearsightedness usually develops during childhood and adolescence, and it usually becomes more stable between the ages of 20 and 40. Myopia tends to run in families. A basic eye exam can confirm nearsightedness.
A number sign (#) is used with this entry because of evidence that high myopia with cataract and vitreoretinal degeneration (MCVD) is caused by homozygous mutation in the LEPREL1 gene (P3H2; 610341) on chromosome 3q28. Clinical Features Mordechai et al. (2011) studied a large consanguineous Israeli Bedouin kindred segregating autosomal recessive nonsyndromic severe myopia with variable expressivity of cataract and vitreoretinal degeneration. The 13 affected family members all presented with poor eyesight in childhood, and all had axial myopia, with increased axial lengths ranging between 25.1 mm and 30.5 mm. Eleven patients developed cataracts that were significant enough to warrant surgery in 1 or both eyes, usually in the first or second decade of life. In 3 patients, subluxated lenses were detected, and were associated with cataract in 2 patients and with lens coloboma in 1 patient.
A number sign (#) is used with this entry because of evidence that myopia-23 (MYP23) is caused by homozygous mutation in the LRPAP1 gene (104225) on chromosome 4p16. Description Myopia, or nearsightedness, is a refractive error of the eye. Light rays from a distant object are focused in front of the retina and those from a near object are focused in the retina; therefore distant objects are blurry and near objects are clear (summary by Kaiser et al., 2004). For a discussion of genetic heterogeneity of myopia, see 160700. Molecular Genetics In 3 consanguineous Saudi Arabian families in which multiple sibs, aged 2 to 16 years, had nonsyndromic extreme myopia with spherical equivalents of -17 diopters or greater and subnormal best-corrected visual acuity, Aldahmesh et al. (2013) performed autozygome analysis and identified only 1 interval exclusively shared among all 8 affected individuals. Linkage analysis confirmed the autozygous interval, and exome sequencing revealed 2 homozygous truncating mutations in the LRPAP1 gene: a 1-bp deletion in 1 family (104225.0001) and a 2-bp deletion in the other 2 families (104225.0002).
A number sign (#) is used with this entry because of evidence that myopia-6 (MYP6) is caused by heterozygous mutation in the SCO2 gene (604272) on chromosome 22q13. Description Myopia, or nearsightedness, is a refractive error of the eye. Light rays from a distant object are focused in front of the retina and those from a near object are focused in the retina; therefore distant objects are blurry and near objects are clear (summary by Kaiser et al., 2004). For a discussion of genetic heterogeneity of susceptibility to myopia, see 160700. Mapping Stambolian et al. (2004) sought to identify a myopia susceptibility gene related to mild/moderate myopia, which is a very common disorder particularly in Chinese and Japanese populations (Saw et al., 1996) and Ashkenazi Jews.
Most research is needed concerning HPAI viruses in wild birds. [24] For example, small birds like sparrows and starlings can be infected with deadly H5N1 strains and they can carry the virus from chicken house to chicken house causing massive epidemics among the chickens. [25] However, pigeons do not present a risk as they neither catch nor carry the virus. [26] [27] [28] Avian flu in humans [ edit ] Human to human transmission [ edit ] The WHO believes that another influenza pandemic is as likely to occur at any time since 1968, when the last century's third of three pandemics took place. [29] The WHO describes a series of six phases, starting with the inter-pandemic period, where there are no new influenza virus subtypes detected in humans, and progressing numerically to the pandemic period, where there is efficient and sustained human-to-human transmission of the virus in the general population. [30] At the present moment, we are at phase 3 on the scale, meaning a new influenza virus subtype is causing disease in humans, but is not yet spreading efficiently and sustainably among humans. [29] So far, H5N1 infections in humans are attributed to bird-to-human transmission of the virus in most cases. ... They noted that EPA specifications for public water supplies that the free chlorine residual values be 6 to 8 mg/L per minute would be "more than sufficient" to inactivate H5N1 in the water environment. ^ Source of quotation: Chotani, Rashid A. (2006), "Part 5 of 6: Interventions" (PDF; slide pack) , The Impact of Pandemic Influenza on Public Health , Johns Hopkins Center for Public Health Preparedness , p. 28 ^ physorg.com Reprint from: American Chemical Society; article "Bird flu virus remains infectious up to 600 days in municipal landfills" published May 27th, 2009 ^ A.R. ... Retrieved 2006-09-15 . ^ "Vietnam to unveil advanced plan to fight bird flu" . Reuters. April 28, 2006. [ permanent dead link ] ^ Webster RG, Peiris M, Chen H, Guan Y (January 2006). ... Retrieved 2006-10-27 . ^ Jennifer Schultz (November 28, 2005). "Bird flu vaccine won't precede pandemic" . ... "Murtha eager to speed vaccine" . Tribune-Review. January 28, 2006 . Retrieved 2006-10-27 . [ permanent dead link ] A promising new bird flu vaccine developed by University of Pittsburgh researchers could provide better protection and be made more quickly than other experimental vaccines. ^ a b "Oseltamivir (Tamiflu)" .
"Predictive Factors for Postherpetic Neuralgia Using Ordered Logistic Regression Analysis". The Clinical Journal of Pain . 28 (8): 712–714. doi : 10.1097/AJP.0b013e318243ee01 . ... PMID 11150362 . ^ a b "Antiviral medications for chickenpox" . Archived from the original on 28 December 2010 . Retrieved 27 March 2011 . ^ "Chickenpox in Children Under 12" . ... "Primary Varicella in Adults: Pneumonia, Pregnancy, and Hospital Admissions". Annals of Emergency Medicine . 28 (2): 165–169. doi : 10.1016/S0196-0644(96)70057-4 . ... MedicineNet.com. Archived from the original on 28 July 2006 . Retrieved 18 August 2006 . ^ "Is Necrotizing Fasciitis a complication of Chickenpox of Cutaneous Vasculitis?" ... ISBN 978-0-306-44855-3 . ^ a b Chicken Pox Complications Archived 28 April 2008 at the Wayback Machine ^ Di Pietrantonj, C; Rivetti, A; Marchione, P; Debalini, MG; Demicheli, V (April 2020).
Overview Chickenpox is an illness caused by the varicella-zoster virus. It brings on an itchy rash with small, fluid-filled blisters. Chickenpox spreads very easily to people who haven't had the disease or haven't gotten the chickenpox vaccine. Chickenpox used to be a widespread problem, but today the vaccine protects children from it. The chickenpox vaccine is a safe way to prevent this illness and the other health problems that can happen during it. Symptoms The rash caused by chickenpox appears 10 to 21 days after you're exposed to the varicella-zoster virus.
Whether symptoms subside after pregnancy is also irrelevant to the diagnosis. [5] A woman is diagnosed with gestational diabetes when glucose intolerance continues beyond 24 to 28 weeks of gestation. The White classification, named after Priscilla White , [6] who pioneered research on the effect of diabetes types on perinatal outcome, is widely used to assess maternal and fetal risk. [7] It distinguishes between gestational diabetes (type A) and pregestational diabetes (diabetes that existed prior to pregnancy). ... The downstream pathways activated include common signaling molecules such as RAS and MAPK, which affect cell motility, cell motility, and cell cycle progression. [28] Studies have shown that HGF is an important signaling molecule in stress related situations where more insulin is needed. ... Future research should include how the method of screening impacts the mother and baby. [35] Pathways [ edit ] Opinions differ about optimal screening and diagnostic measures, in part due to differences in population risks, cost-effectiveness considerations, and lack of an evidence base to support large national screening programs. [36] The most elaborate regimen entails a random blood glucose test during a booking visit, a screening glucose challenge test around 24–28 weeks' gestation, followed by an OGTT if the tests are outside normal limits. ... They are simple to administer and inexpensive, but have a lower test performance compared to the other tests, with moderate sensitivity , low specificity and high false positive rates. [42] [43] [44] Screening glucose challenge test [ edit ] The screening glucose challenge test (sometimes called the O'Sullivan test) is performed between 24–28 weeks, and can be seen as a simplified version of the oral glucose tolerance test (OGTT). ... The risk is highest in women who needed insulin treatment, had antibodies associated with diabetes (such as antibodies against glutamate decarboxylase , islet cell antibodies and/or insulinoma antigen-2 ), women with more than two previous pregnancies, and women who were obese (in order of importance). [79] [80] Women requiring insulin to manage gestational diabetes have a 50% risk of developing diabetes within the next five years. [49] Depending on the population studied, the diagnostic criteria and the length of follow-up, the risk can vary enormously. [81] The risk appears to be highest in the first 5 years, reaching a plateau thereafter. [81] One of the longest studies followed a group of women from Boston, Massachusetts ; half of them developed diabetes after 6 years, and more than 70% had diabetes after 28 years. [81] In a retrospective study in Navajo women, the risk of diabetes after GDM was estimated to be 50 to 70% after 11 years. [82] Another study found a risk of diabetes after GDM of more than 25% after 15 years. [83] In populations with a low risk for type 2 diabetes , in lean subjects and in women with auto-antibodies , there is a higher rate of women developing type 1 diabetes (LADA) . [80] Children of women with GDM have an increased risk for childhood and adult obesity and an increased risk of glucose intolerance and type 2 diabetes later in life. [84] This risk relates to increased maternal glucose values. [85] It is currently unclear how much genetic susceptibility and environmental factors contribute to this risk, and whether treatment of GDM can influence this outcome. [86] Relative benefits and harms of different oral anti-diabetic medications are not yet well understood as of 2017. [73] There are scarce statistical data on the risk of other conditions in women with GDM; in the Jerusalem Perinatal study, 410 out of 37962 women were reported to have GDM, and there was a tendency towards more breast and pancreatic cancer, but more research is needed to confirm this finding. [87] [88] Complications [ edit ] GDM poses a risk to mother and child.
Diagnosis If you're at average risk of gestational diabetes, you'll likely have a screening test during your second trimester — between 24 and 28 weeks of pregnancy. If you're at high risk of diabetes — for example, if you're overweight or obese before pregnancy; you have a mother, father, sibling or child with diabetes; or you had gestational diabetes during a previous pregnancy — your health care provider may test for diabetes early in pregnancy, likely at your first prenatal visit.
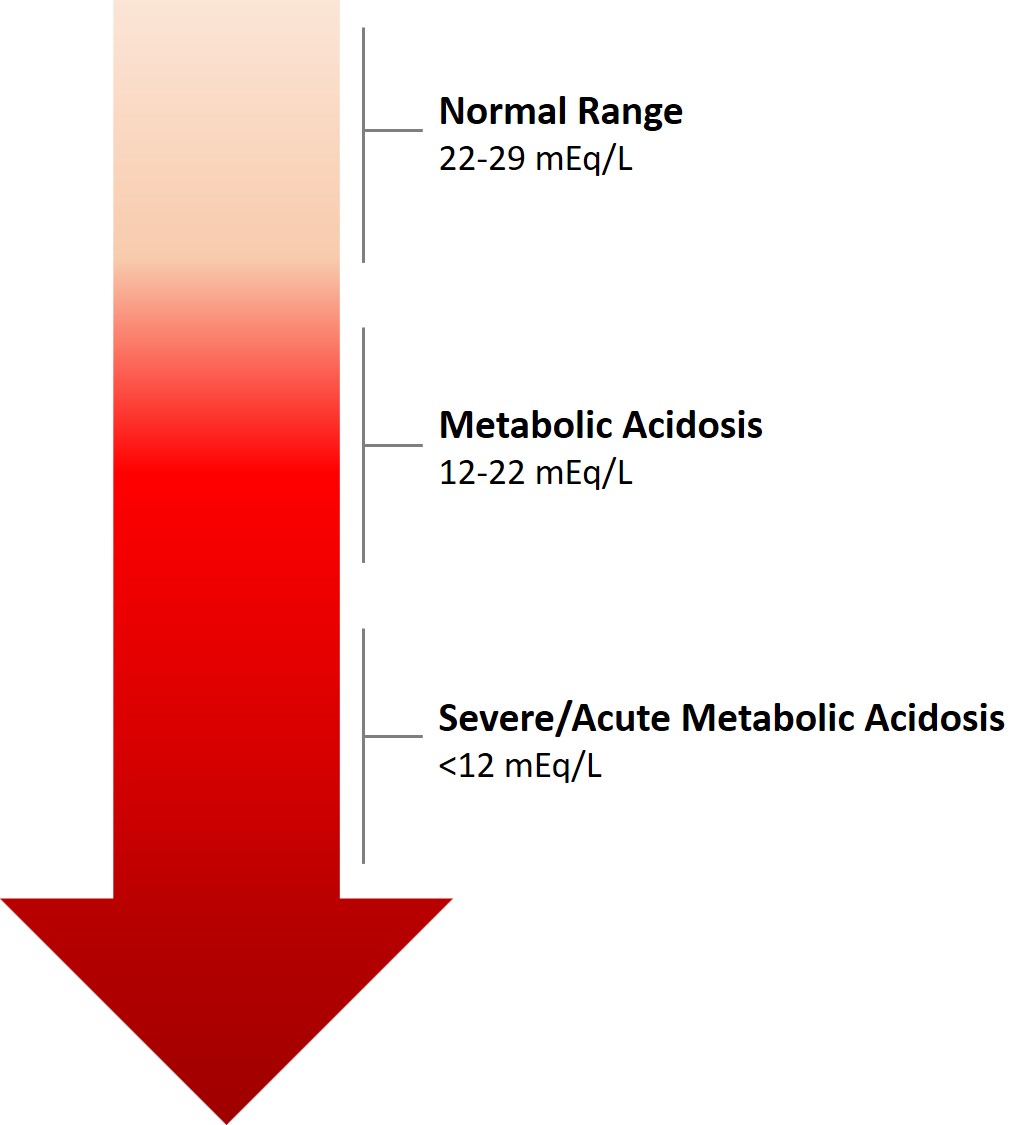
The most adverse consequences of chronic metabolic acidosis in people with Chronic Kidney Disease and in particular, for those who have end-stage renal disease (ESRD) , are detrimental changes to the bones and muscles. [28] Acid buffering leads to loss of bone density, resulting in an increased risk of bone fractures, [29] renal osteodystrophy, [30] and bone disease; [28] as well, increased protein catabolism leads to muscle wasting. [31] [32] Furthermore, metabolic acidosis in Chronic Kidney Disease is also associated with a reduction in eGFR ; it is both a complication of Chronic Kidney Disease, as well as an underlying cause of Chronic Kidney Disease progression. [33] [34] [35] [36] Treatment [ edit ] Treatment of metabolic acidosis depends on the underlying cause, and should target reversing the main process. ... However, amongst the sub-group of patients with severe acute kidney injury, bicarbonate therapy significantly decreased the primary composite outcome, and 28-day mortality, along with the need for dialysis. ... Current Opinion in Nephrology and Hypertension . 28 (3): 267–277. doi : 10.1097/MNH.0000000000000491 . ... Current Opinion in Nephrology and Hypertension . 28 (5): 409–416. doi : 10.1097/MNH.0000000000000524 .
Archived from the original on 18 October 2017 . Retrieved 28 October 2017 . ^ "Final Cumulative Maps and Data | West Nile Virus | CDC" . www.cdc.gov . 24 October 2017. Archived from the original on 27 October 2017 . Retrieved 28 October 2017 . ^ Gompf, Sandra. "West Nile Virus" . ... Retrieved 2018-11-28 . ^ Tyler KL, Pape J, Goody RJ, Corkill M, Kleinschmidt-DeMasters BK (February 2006). ... New York City Department of Health and Mental Hygiene. June 28, 2012. Archived (PDF) from the original on December 3, 2013. ^ Papa A, Karabaxoglou D, Kansouzidou A (October 2011). ... PMID 21837806 . ^ a b "Prevention | West Nile Virus | CDC" . www.cdc.gov . 2018-09-24 . Retrieved 2018-11-28 . ^ American Academy of Pediatrics (8 August 2012).
PMID 30698523 . ^ "Diphtheria—Symptoms—NHS Choices" . Archived from the original on 28 June 2015 . Retrieved 28 June 2015 . ^ a b "Updating PubMed Health" . ... Archived from the original on 17 October 2014 . Retrieved 28 June 2015 . ^ "Diphtheria Symptoms" . www.cdc.gov . 10 April 2017 . ... Economics and Human Biology . 1–1 (1): 1–28. doi : 10.1016/S1570-677X(02)00002-3 . ... Archived from the original on 1 July 2014 . Retrieved 28 October 2013 . ^ Immunology and Vaccine-Preventable Diseases – Pink Book – Diphtheria , CDC ^ Mackie, Elizabeth M; Scott Wilson, T. (12 November 1994). ... "Two cases of deadly diphtheria detected in Lothian area" . ^ "Οκτάχρονος πέθανε από διφθερίτιδα στην Αθήνα - Δεν είχε εμβολιαστεί" . ΑμεΑ Care (in Greek). 28 November 2019 . Retrieved 28 November 2019 .
A rare bacterial infectious disease characterized by an affliction of the upper respiratory tract mediated by the toxin of Corynebacterium diphtheriae . Symptoms include formation of an inflammatory pseudomembrane, fever, sore throat, headaches, coughing, dysphagia, dyspnea, and prominently swollen cervical lymph nodes. The disease may lead to respiratory failure and severe toxin-mediated damage of internal organs, including the heart and kidneys. A cutaneous form of diphtheria is more common in tropical climates and usually follows an indolent course.
Overview Diphtheria (dif-THEER-e-uh) is a serious bacterial infection that usually affects the mucous membranes of the nose and throat. Diphtheria is extremely rare in the United States and other developed countries thanks to widespread vaccination against the disease. However, many countries with limited health care or vaccination options still experience high rates of diphtheria. Diphtheria can be treated with medications. But in advanced stages, diphtheria can damage the heart, kidneys and nervous system. Even with treatment, diphtheria can be deadly, especially in children.
Bonne-Tamir et al. (1990) provided a full analysis of Wilson disease in Israel. From a study of 28 Canadian families, Cox et al. (1972) suggested that there are at least 3 forms of Wilson disease. ... Her sister had died of cirrhosis and liver failure at age 28. Alcohol intake was minimal or completely avoided in all. ... In the first family, the second and fourth male children demonstrated onset of the neurologic type of Wilson disease at 16 and 28 years of age, respectively, and the first female child developed the hepatic type at 38 years of age. ... Strong LD was detected between these markers and the WND locus in 28 families from rural Russia, 43 families from Sardinia, and 67 families of predominantly North American and European descent. ... In 120 unrelated Korean patients with Wilson disease, Park et al. (2007) identified 28 different mutations, including 6 novel mutations, in the ATP7B gene.
Overview Wilson's disease is a rare inherited disorder that causes copper to accumulate in your liver, brain and other vital organs. Most people with Wilson's disease are diagnosed between the ages of 5 and 35, but it can affect younger and older people, as well. Copper plays a key role in the development of healthy nerves, bones, collagen and the skin pigment melanin. Normally, copper is absorbed from your food, and excess is excreted through a substance produced in your liver (bile). But in people with Wilson's disease, copper isn't eliminated properly and instead accumulates, possibly to a life-threatening level.
Wilson disease is an inherited disorder in which excessive amounts of copper accumulate in the body, particularly in the liver, brain, and eyes. The signs and symptoms of Wilson disease usually first appear between the ages of 6 and 45, but they most often begin during the teenage years. The features of this condition include a combination of liver disease and neurological and psychiatric problems. Liver disease is typically the initial feature of Wilson disease in affected children and young adults; individuals diagnosed at an older age usually do not have symptoms of liver problems, although they may have very mild liver disease. The signs and symptoms of liver disease include yellowing of the skin or whites of the eyes (jaundice), fatigue, loss of appetite, and abdominal swelling.
Wilson disease is a very rare inherited multisystemic disease presenting non-specific neurological, hepatic, psychiatric or osseo-muscular manifestations due to excessive copper deposition in the body.
Summary Clinical characteristics. Wilson disease is a disorder of copper metabolism that can present with hepatic, neurologic, or psychiatric disturbances, or a combination of these, in individuals ranging from age three years to older than 50 years; symptoms vary among and within families. Liver disease includes recurrent jaundice, simple acute self-limited hepatitis-like illness, autoimmune-type hepatitis, fulminant hepatic failure, or chronic liver disease. Neurologic presentations include movement disorders (tremors, poor coordination, loss of fine-motor control, chorea, choreoathetosis) or rigid dystonia (mask-like facies, rigidity, gait disturbance, pseudobulbar involvement). Psychiatric disturbance includes depression, neurotic behaviors, disorganization of personality, and, occasionally, intellectual deterioration. Kayser-Fleischer rings, frequently present, result from copper deposition in Descemet's membrane of the cornea and reflect a high degree of copper storage in the body.
Wilson disease is a rare inherited disorder that is characterized by the accumulation of copper in the body. Because high levels of copper are toxic to tissues and organs, this buildup can lead to damage of the liver, brain and eyes. Signs and symptoms of Wilson disease include chronic liver disease, central nervous system abnormalities, and psychiatric (mental health-related) disturbances. It is caused by a mutation of the ATP7B gene and is inherited in an autosomal recessive manner. Although there is no cure for Wilson disease, therapies exist that aim to reduce or control the amount of copper that accumulates in the body.
Liver and neurologic damage that occurs prior to treatment may improve, but it is often permanent. [20] History [ edit ] The disease bears the name of the British physician Samuel Alexander Kinnier Wilson (1878–1937), a neurologist who described the condition, including the pathological changes in the brain and liver, in 1912. [21] Wilson's work had been predated by, and drew on, reports from German neurologist Carl Westphal (in 1883), who termed it "pseudosclerosis"; by the British neurologist William Gowers (in 1888); [22] by the Finnish neuropathologist Ernst Alexander Homén (in 1889–1892), who noted the hereditary nature of the disease; [23] and by Adolph Strümpell (in 1898), who noted hepatic cirrhosis. [22] Neuropathologist John Nathaniel Cumings made the link with copper accumulation in both the liver and the brain in 1948. [24] The occurrence of hemolysis was noted in 1967. [25] In 1951, Cumings, and New Zealand neurologist Derek Denny-Brown , working in the United States, simultaneously reported the first effective treatment, using metal chelator British anti-Lewisite . [26] [27] This treatment had to be injected but was one of the first therapies available in the field of neurology, a field that classically was able to observe and diagnose but had few treatments to offer. [22] [28] The first effective oral chelation agent, penicillamine , was discovered in 1956 by British neurologist John Walshe. [29] In 1982, Walshe also introduced trientine, [30] and was the first to develop tetrathiomolybdate for clinical use. [31] Zinc acetate therapy initially made its appearance in the Netherlands, where physicians Schouwink and Hoogenraad used it in 1961 and in the 1970s, respectively, but it was further developed later by Brewer and colleagues at the University of Michigan . [18] [32] The genetic basis of Wilson's disease, and its linkage to ATP7B mutations, was elucidated by several research groups in the 1980s and 1990s. [33] [34] Other animals [ edit ] Hereditary copper accumulation has been described in Bedlington Terriers , [35] where it generally only affects the liver.
Havener (1951) described macular degeneration with cerebellar ataxia in a 28-year-old black. Cerebellar involvement was much less severe than in a daughter who died at 3 years of age with profound involvement. ... Decreased visual acuity occurred in 83%, with blindness in 28%. Optic atrophy was present in 69%; pigmentary retinopathy in 43%; supranuclear ophthalmoplegia in 56%; and viscosity of eye movements in 79%. ... Stevanin et al. (1998) stated that normal ATXN7 alleles carry from 4 to 35 CAG repeats, whereas pathologic alleles carry from 37 to approximately 200. Intermediate ATXN7 alleles, with 28 to 35 repeats, are exceedingly rare in the general population and are not associated with the SCA7 phenotype, although they were found among relatives of 4 SCA7 patients. In 2 such families, intermediate alleles bearing 35 and 28 CAG repeats gave rise, during paternal transmission, to ATXN7 expansions of 57 and 47 repeats, respectively, that were confirmed by haplotype reconstructions in one case and by inference in the other. ... A second range could be identified with 28 to 35 repeats, and Giunti et al. (1999) provided evidence that these repeats represent intermediate alleles that are prone to further expansion.
Spinocerebellar ataxia 7 (SCA7) is an inherited disease of the central nervous system that leads to impairment of specific nerve fibers carrying messages to and from the brain, resulting in degeneration of the cerebellum (the coordination center of the brain). SCA7 differs from most other forms of SCA in that visual problems, rather than poor coordination, are generally the earliest signs of the disease. Affected individuals have progressive changes in vision (which can result in blindness); symptoms of ataxia ; slow eye movements; and mild changes in sensation or reflexes. Later symptoms include loss of motor control, unclear speech (dysarthria), and difficulty swallowing (dysphagia). Onset in early childhood or infancy has an especially rapid and aggressive course often associated with failure to thrive and regression of motor milestones.
An autosomal dominant cerebellar ataxia type II that is characterized by progressive ataxia, motor system abnormalities, dysarthria, dysphagia and retinal degeneration leading to progressive blindness. Epidemiology The disorder is estimated worldwide prevalence is less than 1/100,000 and it is thought to account for 2-4% of all forms of the disease(up to 7% in Asian populations). Higher prevalence is described in some populations such as in Scandinavia or South Africa. Clinical description Onset of Spinocerebellar ataxia type 7 (SCA7) is generally in the second to fourth decade but can range from infancy to the sixth decade of life. Manifestations that present in infancy and early childhood include muscle weakness, wasting, hypotonia, poor feeding, failure to thrive and loss of motor milestones.
These tests are easily transported and can be performed by people without special training. [28] They are useful for screening large numbers of people and testing people who cannot access healthcare facilities, but their sensitivity is relatively low, [2] and it is recommended that a second method is used to confirm a positive result. [28] [29] T. cruzi can be isolated from samples through blood culture or xenodiagnosis , or by inoculating animals with the person's blood. ... Xenodiagnosis involves feeding the person's blood to triatomine insects, then examining their feces for the parasite 30 to 60 days later. [28] These methods are not routinely used, as they are slow and have low sensitivity. [27] [28] Prevention [ edit ] Bed nets can be used in endemic areas to prevent bites from triatomine bugs. [15] Efforts to prevent Chagas disease have largely focused on vector control to limit exposure to triatomine bugs. ... Eleven triatomine species are native to the United States and some southern states have persistent cycles of disease transmission between insect vectors and animal reservoirs, [2] [23] which include woodrats, possums, raccoons, armadillos and skunks. [49] However, locally acquired infection is very rare: only 28 cases were documented from 1955 to 2015. [2] [48] As of 2013, the cost of treatment in the United States was estimated to be US$900 million annually (global cost $7 billion), which included hospitalization and medical devices such as pacemakers. [40] Chagas disease affects approximately 68,000 to 123,000 people in Europe as of 2019. [50] Spain, which has a high rate of immigration from Latin America, has the highest prevalence of the disease. ... Archived from the original on 4 December 2014 . Retrieved 28 November 2014 . ^ a b c d e f g h i j k Despommier DD, Griffin DO, Gwadz RW, Hotez PJ, Knirsch CA (2019).
Overview Chagas (CHAH-gus) disease is an inflammatory, infectious disease caused by the parasite Trypanosoma cruzi. This parasite is found in the feces of the triatomine (reduviid) bug. This bug is also known as the "kissing bug." Chagas disease is common in South America, Central America and Mexico, the primary home of the triatomine bug. Rare cases of Chagas disease have also been found in the southern United States. Also called American trypanosomiasis, Chagas disease can infect anyone.
A tropical disease mainly found in latin America and transmitted by triatomine insects (mostly Triatoma infestans and Rhodnius prolixus and Panstrongylus megistus ) harboring the hemoflagellate protozoan parasite Trypanosoma cruzi . The disease is characterized by an acute phase which is either asymptomatic or manifest with fever, inflammation at the inoculation site (inoculation chancre or chagoma), unilateral palpebral edema called the Romaña sign (when the triatomine bite occurs near the eye), enlarged lymph nodes, and splenomegaly. The chronic phase is lifelong and development of chagasic cardiomyopathy (30%; complex arrhythmias, heart failure, and thromboembolic events), digestive (10%; megaoesophagus and megacolon), neurological (10%; stroke, peripheral neuropathy and autonomic dysfunction), or mixed alterations (10%) may be observed. These can all lead to high morbidity and mortality rates.
Anterior polar cataracts are small opacities on the anterior surface of the lens, which do not usually interfere with vision. Pras et al. (2006) examined 28 family members from 3 Moroccan Jewish families with autosomal dominant posterior polar cataract (CTPP).
A rare, genetic, non-syndromic developmental defect of the eye disorder, with high clinical and genetic heterogeneity, most frequently characterized by bilateral, symmetrical, non-progressive cataracts which present at birth or in early-childhood. Additional ocular manifestations (e.g. anterior segment dysgenesis, colobomas, nystagmus, microcornea, microphthalmia, myopia) may be associated, however other organs/systems are usually not affected.
Description Anterior polar cataracts are small opacities on the anterior surface of the lens. They usually do not interfere with vision (Moross et al., 1984). The preferred title/symbol of this entry was formerly 'Cataract, Anterior Polar, 2; CTAA2.' Mapping By genetic linkage analysis with microsatellite markers in a 4-generation pedigree, Berry et al. (1996) identified a locus for an autosomal dominant anterior polar cataract on 17p. A maximum lod score of 4.01 at theta = 0.05 was obtained for marker D17S849 and a maximum lod score of 4.17 at theta = 0.05 for D17S796. Multipoint analysis gave a maximum lod score of 5.2. History Hejtmancik (1998) presented a table of 9 loci, including this one, that had been implicated in nonsyndromal cataract and mapped to specific chromosomal sites.
Subsequent screening of KLHL40 by Sanger sequencing in additional probands with severe NEM resulted in the identification of a total of 19 variants in 28 (19.6%) of 143 families affected by severe NEM.
Cushing's disease , a disorder in which cortisol levels are abnormally high Hyperaldosteronism (including Conn's syndrome ), a condition in which aldosterone is over-produced Hypoaldosteronism , a condition in which aldosterone is under-produced References [ edit ] ^ "Overview of the Adrenal Glands: Adrenal Gland Disorders: Merck Manual Home Health Handbook" . Retrieved 2009-03-28 . ^ a b Adrenal Glands , Johns Hopkins Medicine Health Library. ^ Mandel, Lee R.
A more recent account of the Stendhal syndrome was in 2018, where a visitor to the Uffizi Gallery in Florence suffered a heart attack while admiring Sandro Botticelli 's The Birth of Venus . [6] See also [ edit ] Double Rainbow Jerusalem syndrome Lisztomania Paris syndrome The Stendhal Syndrome , a psychological thriller film on the subject References [ edit ] ^ a b Nick Squires (28 July 2010). "Scientists investigate Stendhal Syndrome – fainting caused by great art" .
"The emergence of delusional companions in Alzheimer's disease: an unusual misidentification syndrome". Cogn Neuropsychiatry . 7 (4): 317–28. doi : 10.1080/13546800244000021 .
. — Passer and Warnock, 1991 [11] Diane was a 28-year-old single woman who was seen for an evaluation at a day hospital program in preparation for discharge from a psychiatric hospital. ... Since the patient could not put together memories and feelings, he believed objects in a photograph were new on every viewing, even though they normally should have evoked feelings (e.g., a person close to him, a familiar object, or even himself). [28] Others like Merrin and Silberfarb (1976) [29] have also proposed links between the Capgras syndrome and deficits in aspects of memory. ... "The case of lost Wilma: a clinical report of Capgras delusion". Neurological Sciences . 28 (4): 188–195. doi : 10.1007/s10072-007-0819-8 .